
Амиотрофия шарко код мкб

Связанные заболевания и их лечение
Описания заболеваний
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Классификация
- Симптомы
- Диагностика
- Лечение
Названия
Название: Невральная амиотрофия Шарко-Мари-Тута.
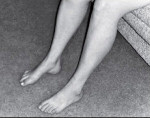
Невральная амиотрофия Шарко-Мари-Тута
Описание
Невральная амиотрофия Шарко. Мари – Тута – прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук. Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц. Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение.
Дополнительные факты
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания. Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования. Лица мужского пола болеют чаще, чем женщины.
По различным данным невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. Населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ.
Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха. В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии. Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ.
Патогенетические аспекты.
На сегодняшний день неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы. Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов. Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону.
Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна. Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути.
Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду.
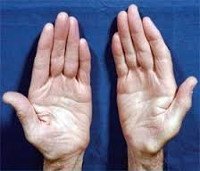
Невральная амиотрофия Шарко-Мари-Тута
Классификация
В современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение. Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса, при ШМТ II типа скорость проведения страдает незначительно. Биопсия нерва обнаруживает при I типе сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток; при II типе — дегенерацию аксонов.
Симптомы
Невральная амиотрофия Шарко-Мари-Тута начинается с развития симметричных мышечных атрофий в дистальных отделах ног. Начальные симптомы манифестируют, как правило, в первой половине второго десятилетия жизни, реже в период от 16 до 30 лет. Они заключаются в повышенной утомляемости стоп при необходимости длительно стоять на одном месте. При этом наблюдается симптом «топтания» – чтобы снять утомляемость стоп пациент прибегает к ходьбе на месте. В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов.
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей.
Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Тремор рук.
Диагностика
Возраст начала заболевания, его типичная клиника, симметричный характер поражения, медленное неуклонное распространение атрофий и усугубляющаяся в связи с этим симптоматика во многих случаях позволяют предположить невральную амиотрофию. Проводимый неврологом осмотр выявляет мышечную слабость в стопах и голенях, деформацию стоп, отсутствие или значительное снижение ахилловых и коленных рефлексов, гипестезию стоп. Для дифференциации ШМТ от других нервно-мышечных заболеваний (миотонии, миопатии, БАС, невропатией) проводится электромиография и электронейрография. С целью исключения метаболической невропатии проводится определение сахара крови, исследование гормонов щитовидной железы, тест на наркотики.
Всем пациентам для уточнения диагноза рекомендована консультация генетика и проведение ДНК-диагностики. Последняя не дает 100% точного результата, т. Пока известны не все генетические маркеры ШМТ. Более точным способом диагностики является введенное в 2010г. Секвенирование генома. Однако это исследование пока является слишком дорогостоящим для широкого использования.
Иногда возникают затруднения в дифференциальной диагностике болезни Шарко-Мари-Тута с невритом Дежерина – Сотта, дистальной миопатией Гоффманна, хроническими полиневропатиями. В таких случаях может потребоваться биопсия мышц и нервов.
Лечение
На современном этапе радикальные способы лечения генных болезней не разработаны. В связи с этим применяется симптоматическая терапия. Проводятся повторные курсы внутримышечного введения витаминов группы В и витамина Е. С целью улучшения мышечной трофики используют АТФ, инозин, кокарбоксилазу, глюкозу. Назначаются ингибиторы холинэстеразы (неостигмин, оксазил, галантамин), препараты для улучшения микроциркуляции и антиоксиданты (никотиновая кислота, пентоксифиллин, мельдоний).
Наряду с фармакотерапией по рекомендации физиотерапевта активно применяются физиотерапевтические методики: электрофорез, СМТ, электростимуляция, диадинамотерапия, грязелечение, УЗ-терапия, оксигенобаротерапия. Целесообразно водолечение сероводородными, сульфидными, хвойными, радоновыми лечебными ваннами. Большое значение в сохранении двигательной активности пациентов, предотвращении развития деформаций и контрактур имеют ЛФК и массаж. При необходимости ортопедом назначается ортопедическое лечение.
Источник
Болезнь Шарко (в литературе еще встречается как «Болезнь Шарко-Мари», «Болезнь Шарко-Мари-Тута», код по МКБ- G60) –генетически обусловленное хроническое заболевание нервной системы, которое характеризуется развитием постоянно прогрессирующей периферической полинейропатией.
Частота диагностирования болезни Шарко-Мари-Тута согласно статистическим данным где-то один случай на две с половиною тысячи пациентов. Первые симптомы появляются в молодом возрасте. Выраженность симптомов и скорость прогрессирования болезни Шарко-Мари разная у каждого пациента. Процент инвалидизации при заболевании очень высок.

Причины заболевания болезнью Шарко:
- Мутация гена PMP22;
- Мутация MPZ;
- Мутация GJB1;
- Мутация MFN и др.
Пути наследования:
- аутосомно-доминантный (чаще всего);
- аутосомно-рецессивный;
- Х-сцепленный.
Заболевание имеет много форм, вызванные разным видом мутаций. Качество жизни и возможности работоспособности при болезни Шарко-Мари-Тута значительно ухудшаются, но на продолжительности жизни обычно это не сказывается.
Симптомы болезни Шарко связанные с поражением моторных и сенсорных нервных волокон. Диагностика болезни Шарко-Мари заключается в исключении диагнозов, которые могут давать подобную клиническую картину, и в проведении ДНК-диагностики, но учитывая, что не все виды мутаций известны, она не всегда информативна.
Лечение болезни Шарко-Мари-Тута заключается в симптоматической терапии. Специфическое лечение на данный момент все еще находится в стадии разработки.
Юсуповская больница – одно из лучших медицинских учреждений, где лечатся пациенты с болезнью Шарко-Мари-Тута. Несмотря на то, что терапия направлена только на купирование симптомов, неврология стремительно развивается и питается найти способы лечения многих заболеваний, в том числе и болезни Шарко. Ведение пациентов изданной патологией достаточно сложное, ведь клиническая картина разнообразна, симптомы выражены в неодинаковой степени и п.т. Опыт работы специалистов Юсуповской больницы позволяет оказывать качественную и эффективную медицинскую помощь. Доктора следят за клиническими исследованиями, новыми разработками, препаратами, изучают их эффективность. В случае необходимости, диагностика проводится быстро и с использованием нового оборудования. Персонал работает на благо пациента.
Симптомы болезни Шарко
Симптомы болезни Шарка-Мари появляются в молодом возрасте, чаще всего до двадцати лет. Прогрессирует заболевание постепенно, пациенты долгое время сохраняют работоспособность и возможность самообслуживания. Причинами, которые ускоряют развитие заболевания, могут стать вирусные и бактериальные инфекции, воздействия неблагоприятных факторов среды, травматизм, недостаток витаминов и т.п.
По МКБ-10 болезнь Шарко-Мари-Тута кодируется как «G60 Наследственная моторная и сенсорная невропатия», что связано в какой-то мере с клинической картиной.
В начале заболевания беспокоит чрезмерная усталость, невозможность долго стоять на одном месте, чувство онемения, «беганье мурашек», а дальше присоединяется атрофическое поражение мышц стоп, которое чаще носит симметричный характер. При обследовании выявляют выпадение сухожильных рефлексов. Стопа деформируется на столько, что больные не могут ходить, опираясь на пятки. Прогрессируя, атрофический процесс поражает голени и бедра.
Дальнейшее развитие заболевания болезни Шарко-Мари-Тута приводит к втягиванию в процесс и кистей, далее – предплечий. Мышцы шеи, туловища и плечевого пояса не атрофируются. Мышцы проксимальных отделов компенсаторно увеличиваются.
Все виды чувствительности нарушаются, но больше всего страдает поверхностная, особенно температурная.
Диагностика болезни Шарко
Диагностика включает:
- Осмотр неврологом;
- Лабораторные исследования;
- Инструментальные исследования;
- ДНК-исследования.
Полное обследование необходимо для исключения заболеваний, в которых клиническая картина сходна. К ним относят: боковой амиосклероз, миотония, метаболическая невропатия. Для исключения хронических полинейропатий проводят биопсию мышц.
Лечение болезни Шарко
Все способы лечения болезни Шарко-Мари-Тута не радикальны. Симптоматическое лечение включает медикаментозную терапию, физиотерапию, лечение у ортопеда и т.п.
Физиотерапевтическое лечение болезни Шарко-Мари-Тута включает ЛФК, массаж, электрофорез, диадинамотерапию, терапию лечебными грязями, разные виды ванн и др.
Медикаментозная терапия направлена на улучшение питания мышечных волокон. С этой целью назначают кокарбоксилазу, глюкозу, аденозинтрифосфат и др. Так же широко применяют антиоксидантные средства, препараты, улучшающие микроциркуляцию, и витамины. Хорошо зарекомендовали себя препараты, которые тормозят активность ацетилхолинэстеразы и повышают уровень ацетилхолина, например, прозерин, галантамин.
Дальнейшие разработки новых препаратов, направленные на радикальные меры – это мир без болезни Шарко-Мари-Тута. Прогрессирование болезни Шарко-Мари-Тута не отражается на том, сколько живут пациенты.
В Юсуповской больнице специалисты долгие годы помогают пациентам держать болезнь под контролем. Минимальная выраженность симптомов и медленное прогрессирование – результат работы врачей. В комфортных палатах, на новых тренажерах, в хорошо оснащенных кабинетах – вот где проходит лечение болезни Шарко-Мари-Тута. Не затягивайте с лечением, запишитесь на консультацию.
Автор

Заведующий отделением восстановительной медицины, врач по лечебной физкультуре, невролог, рефлексотерапевт
Список литературы
- МКБ-10 (Международная классификация болезней)
- Юсуповская больница
- Батуева Е.А., Кайгородова Н.Б., Каракулова Ю.В. Влияние нейротрофической терапии не нейропатическую боль и психовегетативный статус больных диабетической нейропатией // Российский журнал боли. 2011. № 2. С. 46.
- Бойко А.Н., Батышева Т.Т., Костенко Е.В., Пивоварчик Е.М., Ганжула П.А., Исмаилов А.М., Лисинкер Л.Н., Хозова А.А., Отческая О.В., Камчатнов П.Р. Нейродикловит: возможность применения у пациентов с болью в спине // Фарматека. 2010. № 7. С. 63–68.
- Морозова О.Г. Полинейропатии в соматической практике // Внутренняя медицина. 2007. № 4 (4). С. 37–39.
Наши специалисты

Заведующий отделением неврологии, врач-невролог, к.м.н.

Врач-невролог, к.м.н.
Заведующий отделением восстановительной медицины, врач по лечебной физкультуре, невролог, рефлексотерапевт

Врач-невролог

Врач-невролог

Врач-невролог, ведущий специалист отделения неврологии

Врач-невролог

Врач-невролог, к.м.н.
Цены на услуги *
*Информация на сайте носит исключительно ознакомительный характер. Все материалы и цены, размещенные на сайте, не являются публичной офертой, определяемой положениями ст. 437 ГК РФ. Для получения точной информации обратитесь к сотрудникам клиники или посетите нашу клинику.
Скачать прайс на услуги
Мы работаем круглосуточно
Источник
Болезнь Шарко-Мари-Тута, или невральная амиотрофия – наследственная нейропатия с поражением периферической нервной системы. Заболевание получило название по именам трех ее исследователей: французских врачей Жана-Мартина Шарко и Пьера Мари и английского доктора Говарда Генри Тута. В литературе встречается вариант с фамилиями без дефисов, а также краткий вариант – болезнь Шарко.
Еще одно наименование данной патологии – наследственная моторно-сенсорная нейропатия – отражает характер поражения функции нервов. Страдает как моторная (двигательная) функция, так и сенсорная – чувствительность. Код заболевания по классификации МКБ-10 – G60.0.
Предполагается, что изучены не все случаи заболевания, поскольку приблизительно у 10-15% больных симптомы не сильно выражены. Эти люди не знают о своей болезни либо соотносят ее с уже имеющимися другими расстройствами здоровья – например, нейропатией при сахарном диабете. Особенно это характерно для пациентов зрелого возраста, когда состояние здоровья уже не идеально.

Симптомы
Клиническая картина болезни имеет общие симптомы независимо от типа, но при этом может проявляться индивидуально. Даже в одной семье, когда заболевание провоцируется одним и тем же геном, у двух близких родственников оно далеко не всегда проявляется одинаково.
Общие симптомы болезни Шарко:
- атрофия мышц дистальных отделов конечностей (голень, предплечье);
- появление нечувствительных участков, онемение (без «мурашек» и покалываний);
- нарушение рефлексов;
- увеличение свода стопы (утолщение голеностопного сустава);
- сколиоз, кифоз, другие деформации скелета.

Дополнительные симптомы при 1-м типе болезни:
- сильные судороги в мышцах голени (чаще в передних отделах) после физической нагрузки;
- нарастающая слабость мышц;
- изменение походки, у детей – ходьба на цыпочках;
- нарушение тонуса мышц-сгибателей и разгибателей;
- молоткообразная форма пальцев ног (вместе с увеличением свода стопы);
- атрофия мышц ног, начиная с дистальных (дальних) отделов – от стопы к колену; «аистообразная», «бутылкообразная» форма ног;
- по мере развития болезни – дрожание рук, слабость пальцев, нарушение мелкой моторики;
- нарушение сухожильных и периостальных рефлексов в области кистей и стоп;
- нарушение вибрационной, тактильной, а затем термальной и болевой чувствительности кистей и стоп;
- утолщение нервных стволов;
- искривления позвоночника;
- возможна атипичная форма болезни в виде синдрома Руси-Леви: топтание при попытке стоять неподвижно, неустойчивость при ходьбе, сильное дрожание рук в статичном положении, ночные судороги в икроножных мышцах, парезы.

Особенности 2-го типа заболевания:
- изменения подъема стопы и формы пальцев встречаются реже, чем при 1-м типе;
- не наблюдаются утолщения нервных стволов;
- меньшая степень нарушения чувствительности;
- наблюдается синдром беспокойных ног перед сном;
- реже встречается ослабевание мышц в руках;
- возможно нарушение слуха (когда болезнь передается по женской линии);
- транзисторная энцефалопатия после физической активности на высоте: нарушения речи, шатание при ходьбе, затрудненное глотание, слабость проксимальных (ближних к туловищу) отделах конечностей.
Проявлением данной патологии обоих типов могут стать аутоиммунные реакции, когда вырабатываемые организмом специальные антитела разрушают миелиновые оболочки нервных волокон.

Причины болезни Шарко
В основе механизма развития болезни лежит мутация генов. Однако первопричину этого явления наука пока не открыла, хотя патология описана еще в 19 веке. Происхождение невральной амиотрофии, ее истинные причины все еще не изучены до конца. Для полноты информации ученым потребуется изучить все возможные генные изменения и их механизмы. Сегодня ученые выявили около 20 генов, в которых происходят точечные мутации, которые приводят к развитию болезни Шарко-Мари-Тута. Чаще всего это гены PMP22, MPZ, MFN2 и некоторые другие. Интересно, что чаще других затрагиваются генные структуры 1-й и 8-й хромосом. О многих других генах до сих пор достоверно ничего не известно.
Существует даже предположение, что открыты не все разновидности заболевания. Ниже речь пойдет о двух основных его типах, наиболее часто встречающихся. Однако современной медицине известно не менее 4 видов невральной амиотрофии.
Некоторые факторы могут спровоцировать развитие болезни Шарко при имеющейся генетической предрасположенности. Это употребление алкогольных напитков, нездоровый образ жизни, использование потенциально вредных для нервной системы лекарственных препаратов. Даже некоторые средства, применяемые для наркоза при операциях (тиопентал), могут стать триггерами – провокаторами патологического процесса, обусловленного генетикой.

Виды заболевания
У 60% пациентов диагностируется болезнь Шарко 1-го, демиелинизирующего типа, которая проявляется в раннем детском возрасте или у молодых людей до 20 лет. У 20% больных обнаруживается 2-й тип этого заболевания – аксональный. Его симптомы, наоборот, начинаются в позднем периоде жизни – в 60-70 лет. Оба типа заболевания передаются аутосомно-доминантным путем. Для заболевания человека достаточно того, чтобы хотя бы один из его родителей страдал болезнью Шарко Мари Тута. Изредка встречается и рецессивная передача патологического гена. В этом случае оба родителя здоровы, но являются носителями заболевания.
У 1-го и 2-го типа различается не только возраст дебюта симптоматики, но и характер повреждений нервной ткани. При демиелинизуриющем типе повреждается миелиновая оболочка нерва, а при аксональном разрушение касается аксона – длинного отростка нейрона.

Что нужно для подтверждения диагноза
Существует несколько различных видов нейропатии, обусловленных разными причинами. Но болезнь Шарко Мари Тута имеет существенное отличие – она передается генетически. Поэтому для точности в постановке диагноза важно исследовать фактор наследственности.
Врач-невролог обязательно изучит не только жалобы пациента, данные аппаратных обследований и лабораторных анализов, но и наличие данного заболевания в семье пациента. Семейный анамнез крайне важен в диагностике.
Для диагностики проводятся:
- сбор и анализ неврологических симптомов;
- электронейромиография (для измерения скорости импульсов и биоэлектрической активности);
- генетический анализ, исследование ДНК;
- осмотр пациента на наличие деформаций костей рук и ног, позвоночника;
- исследование образцов нервной и мышечной тканей (для дифференциации с нейропатиями другого генеза).

Методы лечения
Лечение болезни Шарко направлено на уменьшение проявления симптомов. Специальной терапии, которая бы избавила больного от заболевания навсегда, в настоящее время не существует. Предотвратить болезнь невозможно, поскольку она передается генетически.
 Терапевтические методы включают в себя применение препаратов, улучшающих кровообращение и обменные процессы в тканях, коэнзим Q, витамин Е. Для улучшения нервной проводимости популярное лекарство – Галантамин. При болях в ногах назначаются антидепрессанты, противосудорожные препараты. Опытным путем установлена эффективность больших доз витамина С при 1-м типе болезни. Благодаря использованию витамина С удавалось замедлить разрушение миелиновых оболочек нейронов.
Терапевтические методы включают в себя применение препаратов, улучшающих кровообращение и обменные процессы в тканях, коэнзим Q, витамин Е. Для улучшения нервной проводимости популярное лекарство – Галантамин. При болях в ногах назначаются антидепрессанты, противосудорожные препараты. Опытным путем установлена эффективность больших доз витамина С при 1-м типе болезни. Благодаря использованию витамина С удавалось замедлить разрушение миелиновых оболочек нейронов.
При ухудшении состояния больного (чаще всего из-за включения аутоиммунных симптомов) назначаются иммуноглобулины, кортикостероиды (например, Метилпреднизолон). Может проводиться плазмоферез.
 Для лечения данного заболевания широко применяются ЛФК, физиотерапевтические методы: массаж, гидромассаж, грязевые ванны, бальнеотерапия и другие. При нарушении сенсорных функций нужно осторожно применять гальванизацию и электрофорез. Полезны занятия плаванием, спортивной ходьбой, но без слишком больших нагрузок.
Для лечения данного заболевания широко применяются ЛФК, физиотерапевтические методы: массаж, гидромассаж, грязевые ванны, бальнеотерапия и другие. При нарушении сенсорных функций нужно осторожно применять гальванизацию и электрофорез. Полезны занятия плаванием, спортивной ходьбой, но без слишком больших нагрузок.
В случаях, когда пациент теряет способность самостоятельно передвигаться, может проводиться хирургическая операция. Чаще всего это артродез голеностопных суставов – сращивание большеберцовой и таранной костей. Это придает ноге неподвижность в голеностопе, но исключает опрокидывание при ходьбе и дает человеку возможность ходить.
Симптомы болезни Шарко Мари Тута медленно прогрессируют, но заметно это не всегда. Ведь порой это происходит на протяжении десятилетий. Важно, что наличие данного заболевания не влияет на продолжительность жизни человека – только на ее качество и самочувствие больного. При правильно и своевременно проведенных лечебных процедурах развитие болезни можно замедлить.
Широко применяются в лечении больных различные ортопедические приспособления. Для большей устойчивости голеностопа показано ношение высокой обуви, специальных повязок, которые предупреждают вывихи и растяжения – типичные осложнения этого заболевания.
Однако нужно помнить, что запущенная стадия болезни Шарко приводит к инвалидности. При дистрофических процессах в руках больной утрачивает способность выполнять домашние дела, работу руками, не может себя обслуживать. Если страдают мышцы и суставы ног, для передвижения человеку нужны инвалидная коляска, поддерживающие устройства и опоры.
Тем не менее, большинство заболевших адаптируется к жизни, могут себя обслуживать, передвигаться самостоятельно, даже вести трудовую деятельность. Учитывая возможные проблемы в будущем с движениями, координацией, мелкой моторикой, иногда со слухом, надо постараться заранее ориентироваться (либо ориентировать пациента-ребенка) на выбор подходящей профессии.
Источник