
Аплазия кисти код мкб

Q65 Врожденные деформации бедра
- Q65.0 Врожденный вывих бедра односторонний
- Q65.1 Врожденный вывих бедра двусторонний
- Q65.2 Врожденный вывих бедра неуточненный
- Q65.3 Врожденный подвывих бедра односторонний
- Q65.4 Врожденный подвывих бедра двусторонний
- Q65.5 Врожденный подвывих бедра неуточненный
- Q65.6 Неустойчивое бедро
- Q65.8 Другие врожденные деформации бедра
- Q65.9 Врожденная деформация бедра неуточненная
Q66 Врожденные деформации стопы
- Q66.0 Конско-варусная косолапость
- Q66.1 Пяточно-варусная косолапость
- Q66.2 Варусная стопа
- Q66.3 Другие врожденные варусные деформации
- Q66.4 Пяточно-вальгусная косолапость
- Q66.5 Врожденная плоская стопа [pes planus]
- Q66.6 Другие врожденные вальгусные деформации стопы
- Q66.7 Полая стопа [pes cavus]
- Q66.8 Другие врожденные деформации стопы
- Q66.9 Врожденная деформация стопы неуточненная
Q67 Врожденные костно-мышечные деформации головы, лица, позвоночника и грудной клетки
- Q67.0 Асимметрия лица
- Q67.1 Сдавленное лицо
- Q67.2 Долихоцефалия
- Q67.3 Плагиоцефалия
- Q67.4 Другие врожденные деформации черепа, лица и челюсти
- Q67.5 Врожденная деформация позвоночника
- Q67.6 Впалая грудь
- Q67.7 Килевидная грудь
- Q67.8 Другие врожденные деформации грудной клетки
Q68 Другие врожденные костно-мышечные деформации
- Q68.0 Врожденная деформация грудиноключично-сосцевидной мышцы
- Q68.1 Врожденная деформация кисти
- Q68.2 Врожденная деформация колена
- Q68.3 Врожденное искривление бедра
- Q68.4 Врожденное искривление большеберцовой и малоберцовой костей
- Q68.5 Врожденное искривление длинных костей голени неуточненное
- Q68.8 Другие уточненные врожденные костно-мышечные деформации
Q69 Полидактилия
- Q69.0 Добавочный палец пальцы
- Q69.1 Добавочный большой палец пальцы кисти
- Q69.2 Добавочный палец пальцы стопы
- Q69.9 Полидактилия неуточненная
Q70 Синдактилия
- Q70.0 Сращение пальцев кисти
- Q70.1 Перепончатость пальцев кисти
- Q70.2 Сращение пальцев стопы
- Q70.3 Перепончатость пальцев стопы
- Q70.4 Полисиндактилия
- Q70.9 Синдактилия неуточненная
Q71 Дефекты, укорачивающие верхнюю конечность
- Q71.0 Врожденное полное отсутствие верхней их конечности ей
- Q71.1 Врожденное отсутствие плеча и предплечья при наличии кисти
- Q71.2 Врожденное отсутствие предплечья и кисти
- Q71.3 Врожденное отсутствие кисти и пальца (ев)
- Q71.4 Продольное укорочение лучевой кости
- Q71.5 Продольное укорочение локтевой кости
- Q71.6 Клешнеобразная кисть
- Q71.8 Другие дефекты, укорачивающие верхнюю ие конечность (и)
- Q71.9 Дефект, укорачивающий верхнюю конечность, неуточненный
Q72 Дефекты, укорачивающие нижнюю конечность
- Q72.0 Врожденное полное отсутствие нижней их конечности (ей)
- Q72.1 Врожденное отсутствие бедра и голени при наличии стопы
- Q72.2 Врожденное отсутствие голени и стопы
- Q72.3 Врожденное отсутствие стопы и пальца (ев) стопы
- Q72.4 Продольное укорочение бедренной кости
- Q72.5 Продольное укорочение большеберцовой кости
- Q72.6 Продольное укорочение малоберцовой кости
- Q72.7 Врожденное расщепление стопы
- Q72.8 Другие дефекты, укорачивающие нижнюю (ие) конечность (и)
- Q72.9 Дефект, укорачивающий нижнюю конечность, неуточненный
Q73 Дефекты, укорачивающие конечность неуточненную
- Q73.0 Врожденное отсутствие конечности (ей) неуточненной (ых)
- Q73.1 Фокомелия конечности (ей) неуточненной (ых)
- Q73.8 Другие дефекты, укорачивающие конечность (и) неуточненную (ые)
Q74 Другие врожденные аномалии [пороки развития] конечности (ей)
- Q74.0 Другие врожденные аномалии верхней конечности (ей), включая плечевой пояс
- Q74.1 Врожденная аномалия коленного сустава
- Q74.2 Другие врожденные аномалии нижней их конечности (ей), включая тазовый пояс
- Q74.3 Врожденный множественный артрогрипоз
- Q74.8 Другие уточненные врожденные аномалии конечности (ей)
- Q74.9 Врожденная аномалия конечности (ей) неуточненная
Q75 Другие врожденные аномалии [пороки развития] костей черепа и лица
- Q75.0 Краниосиностоз
- Q75.1 Краниофациальный дизостоз
- Q75.2 Гипертелоризм
- Q75.3 Макроцефалия
- Q75.4 Челюстно-лицевой дистоз
- Q75.5 Окуломандибулярный дистоз
- Q75.8 Другие уточненные пороки развития костей черепа и лица
- Q75.9 Врожденная аномалия костей черепа и лица неуточненная
Q76 Врожденные аномалии [пороки развития] позвоночника и костей грудной клетки
- Q76.0 Spina bifida occulta
- Q76.1 Синдром Клиппеля-Фейля
- Q76.2 Врожденный спондилолистез
- Q76.3 Врожденный сколиоз, вызванный пороком развития кости
- Q76.4 Другие врожденные аномалии позвоночника, не связанные со сколиозом
- Q76.5 Шейное ребро
- Q76.6 Другие врожденные аномалии ребер
- Q76.7 Врожденная аномалия грудины
- Q76.8 Другие врожденные аномалии костей грудной клетки
- Q76.9 Врожденная аномалия костей грудной клетки неуточненная
Q77 Отеоходродисплазия с дефектами роста трубчатых костей и позвоночника
- Q77.0 Ахондрогенезия
- Q77.1 Маленький рост, не совместимый с жизнью
- Q77.2 Синдром короткого ребра
- Q77.3 Точечная хондродисплазия
- Q77.4 Ахондроплазия
- Q77.5 Дистрофическая дисплазия
- Q77.6 Хондроэктодермальная дисплазия
- Q77.7 Спондилоэпифизарная дисплазия
- Q77.8 Другая остеохондродисплазия с дефектами роста трубчатых костей и позвоночного столба
- Q77.9 Остеохондродисплазия с дефектами роста трубчатых костей и позвоночного столба неуточненная
Q78 Другие остеохондродисплазии
- Q78.0 Незавершенный остеогенез
- Q78.1 Полиостозная фиброзная дисплазия
- Q78.2 Остеопетроз
- Q78.3 Прогрессирующая диафизарная дисплазия
- Q78.4 Энхондроматоз
- Q78.5 Метафизарная дисплазия
- Q78.6 Множественные врожденные экзостозы
- Q78.8 Другие уточненные остеохондродисплазии
- Q78.9 Остеохондродисплазия неуточненная
Q79 Врожденные аномалии [пороки развития] костно-мышечной системы, не классифицированные в других рубриках
- Q79.0 Врожденная диафрагмальная грыжа
- Q79.1 Другие пороки развития диафрагмы
- Q79.2 Экзомфалоз
- Q79.3 Гастрошиз
- Q79.4 Синдром сливообразного живота
- Q79.5 Другие врожденные аномалии брюшной стенки
- Q79.6 Синдром Элерса-Данло
- Q79.8 Другие пороки развития костно-мышечной системы
- Q79.9 Врожденный порок развития костно-мышечной системы неуточненный
Источник
Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Врождённая косорукость – комбинированный порок, обусловленный недоразвитием тканей по лучевой или локтевой стороне верхней конечности. При отклонении кисти в лучевую сторону ставят диагноз лучевая косорукость (таnus valga), при отклонении в противоположную сторону – локтевая врождённая косорукость (manus vara).
Код по МКБ-10
- Q71.4 Лучевая врождённая косорукость.
- Q71.5 Локтевая врождённая косорукость.
Чем вызывается врождённая косорукость?
Врождённая косорукость, по данным мировой литературы, регистрируется у 1 из 1400-100 000 детей. Чаще диагностируется лучевая врождённая косорукость. Локтевую наблюдают в 7 раз реже лучевой.
Возникает врождённая косорукость вследствия воздействия внешних и внутренних факторов, которые характерны и для других врождённых аномалий развития верхней конечности. К внешним, экзогенным, факторам относят ионизирующее облучение, механические и психические травмы, лекарственные препараты, контакты с инфекционными заболеваниями, недостаток питания и др. Эндогенные причины – различные патологические изменения и функциональные расстройства матки у беременной, общие заболевания матери, гормональные расстройства, старение организма. При этом большое значение имеют сроки воздействия, а наиболее неблагоприятными для матери считаются первые 4-5 нед беременности. Наследственного фактора не выявлено.
Как проявляется врождённая косорукость?
Врождённая косорукость характеризуется триадой: лучевая девиация кисти (может быть с подвывихом и вывихом кисти в локтезапястном сочленении); недоразвитие костей предплечья (в первую очередь лучевой кости); аномалия развития пальцев и кисти.
Из других поражений кисти возможны гипоплазия и клинодактилия II пальца, синдактилии, сгибательные и разгибательные контрактуры в пястно-фаланговых и межфаланговых суставах, наиболее выраженных во II и III пальцах кисти. Страдают и кости запястья, расположенные по лучевой стороне, при этом наблюдают аплазию или конкресценцию с другими костями.
Классификация
В классификации лучевой косорукости выделяют три степени недоразвития лучевой кости и четыре типа кисти. Основа для классификации – рентгенологическая картина.
[1], [2], [3], [4], [5], [6], [7]
Степени недоразвития лучевой кости
- I степень – укорочение лучевой кости составляет до 50% нормальной её длины.
- II степень – укорочение лучевой кости превышает 50% её нормальной длины.
- III степень – полное отсутствие лучевой кости.
[8], [9], [10]
Типы кисти
Для кисти характерно поражение первого луча (луч – все фаланги пальца и соответствующая пястная кость).
При типе 1 обнаруживают гипоплазию I пястной кости и мышц thenar, тип 2 характеризуется полным отсутствием пястной кости и гипоплазией фаланг I пальца (при этом обычно наблюдают «болтающийся палец»). Тип 3 выражается в аплазии всего первого луча кисти. При типе 4 костные нарушения отсутствуют.
Как лечится врождённая косорукость?
Консервативное лечение
Консервативное лечение (проводят с первых месяцев жизни ребёнка) включает ЛФК, массаж, редрессирующие упражнения для уменьшения существующих контрактур пальцев и кисти, снабжение ортезными изделиями. Однако консервативные мероприятия не дают стойкого положительного результата и их следует рассматривать как предварительную подготовку ко второму этапу – хирургическому. Оперативное лечение рекомендуют начинать с полугодовалого возраста.
[11], [12], [13]
Хирургическое лечение
Выбор метода хирургического воздействия зависит от вида деформации.
Чем меньше степень и чем младше ребёнок, тем легче вывести кисть из девиации. Поэтому рекомендуют начинать хирургическое лечение до 2-3-летнего возраста.
Локтевая врождённая косорукость характеризуется деформацией и укорочением предплечья, локтевой девиацией кисти, ограничением движения в локтевом суставе. В большей степени выражено недоразвитие локтевой кости, особенно её дистального отдела. В этой зоне обычно располагается фиброзно-хрящевой тяж, соединяющий локтевую кость с костями запястья. Лучевая кость дугообразно искривлена. Её головка чаще всего вывихнута в локтевом суставе кпереди и кнаружи, что определяет контрактуру в локтевом суставе. Ось предплечья и кисти отклонена в локтевую сторону. Изменения кисти характеризуются большим разнообразием. Из патологий кисти наиболее часто наблюдают аплазию одного или двух, обычно ульнарных, лучей, а также недоразвитие большого пальца. Из других деформаций сегмента отмечают синдактилии и гипоплазии.
По степени недоразвития локтевой кости выделяется врождённая косорукость 4-х типов.
- Первый вариант – умеренная гипоплазия – длина локтевой кости составляет 61-90% лучевой кости.
- Второй вариант – выраженная гипоплазия – длина локтевой кости составляет 31-60% лучевой кости.
- Третий вариант – рудимент локтевой кости – длина локтевой кости составляет 1-30% лучевой кости.
- Четвёртый вариант – аплазия локтевой кости (полное отсутствие).
Цели и принципы консервативного лечения идентичны целям и принципам лечения лучевой косорукости.
Показанием к оперативному лечению локтевой врожденной косорукости считают невозможность или затруднение самообслуживания аномальной конечностью из-за контрактуры в локтевом суставе (не устраняемой консервативными методами), из-за укорочения предплечья и не корригируемой пассивно локтевой девиации кисти и, наконец, из-за ограничения функции кисти, в первую очередь её двустороннего схвата. Хирургическое вмешательство начинают с ликвидации наиболее функционально значимой деформации. Оперировать можно уже с первого года жизни больного.
В послеоперационном периоде назначают комплекс восстановительных мер, включающих ЛФК, массаж, физиотерапию, направленные на восстановление амплитуды движений и увеличение силы конечностей.
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Прогноз
Названия
Название: Врожденная аплазия кожи.
Врожденная аплазия кожи
Описание
Врожденная аплазия кожи. Группа состояний неясной этиологии, которые характеризуются очаговым нарушением формирования кожных покровов с развитием рубцов. Симптомы этого состояния выявляются сразу после рождения ребенка, у которого наблюдается одна или несколько эрозий или изъязвлений на коже головы или, очень редко, на других участках тела. Диагностика врожденной аплазии кожи осуществляется на основании данных осмотра дерматологом, гистологического изучения тканей в очаге поражения. Лечение только симптоматическое с целью недопущения развития вторичной инфекции, но возможна хирургическая коррекция рубцов для уменьшения косметического дефекта.
Дополнительные факты
Врожденная аплазия кожи – это очаговый дефект развития кожи, при котором нарушается формирование эпидермиса, дермы, придатков, а в особо тяжелых случаях и подкожной клетчатки. Это состояние известно человечеству уже более 250 лет, однако выявить причины его развития до сих пор не удалось, в дерматологии имеются лишь теории на этот счет. Встречаемость врожденной аплазии кожи точно неизвестна, большинство исследователей оценивают ее на уровне 1:10000. Иногда такое состояние сочетается с некоторыми генетическими заболеваниями, другими пороками внутриутробного развития. Врожденная аплазия кожи в большинстве случаев не приводит к тяжелым последствиям, однако косметический дефект в виде рубца на месте патологического очага остается у человека на всю жизнь.
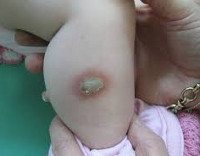
Врожденная аплазия кожи
Причины
На сегодняшний день нет единой и общепризнанной теории, которая бы объясняла развитие этого врожденного дефекта развития кожи. Предполагается, что причиной врожденной аплазии кожи является целая группа различных патологических факторов, которые ведут к нарушению процесса закрытия нервной трубки или же тормозят развитие эмбриональных зачатков дермы и эпидермиса. Иногда удается выявить семейные формы этого состояния, при этом механизм его наследования, предположительно, аутосомно-доминантный. Но намного чаще встречаются спорадические формы врожденной аплазии кожи, причем иногда в сочетании с другими пороками развития, обусловленные генетическими заболеваниями или воздействием тератогенных факторов. Это дает повод рассматривать данное состояние как следствие влияния на формирующийся плод различных повреждающих факторов.
Симптомы
Аплазия кожи выявляется сразу после рождения ребенка. Чаще всего на теменной области обнаруживается очаг округлой формы диаметром 1-3 сантиметра. Примерно в трети случаев возникает два очага, еще реже встречаются три и более участка аплазии кожи. Патологический участок представляет собой эрозию или язву, покрытую корочкой и грануляциями, волосяной покров на нем отсутствует. Однако вокруг язвы вырастают более длинные и темные волоски, что получило название симптома «волосяного воротничка». Цвет образования варьируется от розового до ярко-красного.
Диагностика
Распознавание этого заболевания обычно не представляет труда для врача-дерматолога – его симптоматика достаточно специфична, и спутать его с другими врожденными кожными состояниями довольно сложно. Однако в некоторых случаях похожую на врожденную аплазию кожи картину могут иметь и иные патологические процессы и состояния. Поэтому необходимо производить дифференциальную диагностику этой патологии с такими заболеваниями, как очаговая склеродермия, дискоидная красная волчанка, а также с последствиями перинатальной травмы (от щипцов и других акушерских инструментов). Очень похожи на аплазию семейные формы гипоплазии кожи лица, однако при этом атрофические очаги наблюдаются в области висков.
Наиболее точные диагностические данные может дать гистологическое изучение тканей патологического очага. При врожденной аплазии кожи наблюдается резкое уменьшение толщины (вплоть до 1-го слоя клеток) эпидермиса, дермы, иногда подкожной клетчатки. Признаков воспаления и лейкоцитарной инфильтрации (при отсутствии вторичной инфекции) не наблюдается, также не выявляют придатков кожи.
Лечение
Лечение врожденной аплазии кожи условно разделяется на два этапа. Первый производится непосредственно после рождения ребенка – в этот период показаны только профилактические и уходовые мероприятия (обработка эрозий антибактериальными мазями, увлажняющими средствами), наложение повязки для уменьшения риска травмирования. Через несколько недель на месте патологического очага сформируется рубец, который хоть и остается на всю жизнь, но может быть прикрыт окружающими волосами. Второй этап сводится к хирургическому устранению дефекта (чаще всего, по косметическим причинам), и проводить его можно в позднем детском или взрослом возрасте. При коррекции значительных по площади участков врожденной аплазии кожи может применяться пересадка кожи.
Прогноз
Прогноз заболевания в целом благоприятный, некоторые исследователи указывают на необходимость ежегодного осмотра рубца у дерматолога из-за риска развития онкологических процессов.
Источник
Содержание
- Описание
- Дополнительные факты
- Классификация
- Симптомы
- Диагностика
- Лечение
- Профилактика
Названия
Название: Брахидактилия.
Описание
Брахидактилия. Наследственный дефект, характеризующийся недоразвитием фаланг и укорочением пальцев на руках или ногах. Признаками брахидактилии служит короткопалость, укорочение размера кисти или стопы, гипоплазия ногтевых пластин, часто ригидность суставов (симфалангия), синдактилия, полидактилия. Обследование пациента с брахидактилией включает консультацию генетика, ДНК-диагностику, рентгенографию кистей и стоп. Лечение брахидактилии включает разделение и удлинение пальцев, ЛФК и массаж.
Дополнительные факты
Брахидактилия (гипофалангия, микродактилия) – короткопалость, врожденная аномалия конечностей, выражающаяся в наличии у ребенка коротких пальцев вследствие укорочения или отсутствия отдельных фаланг. Брахидактилия является доминантно-наследуемым признаком, т. Е. Для проявления аномалии у ребенка ген брахидактилии должен быть унаследован от одного из родителей. При отсутствии других аномалий люди с брахидактилией могут жить обычной, нормальной жизнью, однако короткопалость накладывает ограничения на профессиональный выбор. Брахидактилия с одинаковой частотой встречается среди представителей обоих полов; распространенность в популяции – 1,5:100 000.
Классификация
В травматологии и ортопедии различают несколько типов брахидактилии.
Брахидактилию типа А характеризует укорочение средних фаланг пальцев, радиальное искривление фаланг, дисплазия ногтевых пластин.
При брахидактилии типа А1 (типе Фараби) отмечается рудиментарное строение средних фаланг, иногда – их слияние с концевыми фалангами. Имеет место укорочение проксимальных фаланг первых пальцев кистей и стоп, задержка роста.
Для брахидактилии типа А2 (брахимезофалангии, типа Мора-Брита) свойственно укорочение средних фаланг вторых пальцев кистей и стоп; при этом остальные пальцы относительно сохранны. Средние фаланги вторых пальцев имеют треугольную или ромбовидную форму и радиальное отклонение.
При брахидактилии типа А3 (брахимезофалангии V пальца, клинодактилии) наблюдается укорочение и радиальное искривление средних фаланг пятых пальцев кистей.
Брахидактилии типа А4 (брахимезофалангии II и V пальцев, типу Темтами) присуще поражение средних фаланг вторых и пятых пальцев кисти.
При брахидактилии типа А5 отсутствуют средние фаланги вторых-пятых пальцев и имеется дисплазия ногтей.
Брахидактилия типа В характеризуется не только уменьшением длины средних фаланг, но также недоразвитием или полным отсутствием дистальных (концевых) фаланг и синдактилией (сращением) второго и третьего пальцев.
Брахидактилия типа С отличается укорочением проксимальных и средних фаланг второго и третьего пальцев. Данный тип брахидактилии часто сопровождается симфалангией (сращением фаланг), укорочением пястных костей. Может сочетаться с низкорослостью и умственной отсталостью.
Брахидактилия типа D (брахимегалодактилия) диагностируется при укорочении первых пальцев кистей и стоп.
Брахидактилия типа Е характеризуется укорочением пястных и плюсневых костей (костей кисти и стопы).
Наиболее часто в клинической практике встречается брахидактилия типов А3 и D.
Симптомы
Брахидактилия может являться изолированной врожденной аномалией, однако в некоторых случаях короткопалость сопутствует другим врожденным синдромам. Так, при синдроме Дауна, наряду с брахидактилией, имеются следующие характерные признаки: брахицефалия, короткая шея, эпикантус, катаракта, аномалии зубных рядов, бороздчатый язык, врожденные пороки сердца, косоглазие, деформация грудной клетки (воронкообразная или килевидная) и тд.
Минимальными диагностическими признаками синдрома Беймонда служат брахидактилия, нистагм, мозжечковая атаксия. При синдроме Поланда отсутствуют большая и малая грудные мышцы; имеет место брахидактилия, синдактилия, аплазия сосков или амастия, деформация ребер и тд.
Синдром срединной расщелины лица характеризуется микрогенией, атрезией хоан, гипоплазией лобных пазух, брахидактилией, гипофизарным нанизмом, вторичным гипотиреозом, двусторонним крипторхизмом, искривлением полового члена, олигофренией и пр.
При синдроме Аарскога-Скотта, кроме брахидактилии, имеет место гипертелоризм, низкорослость, разболтанность суставов, синдактилия, шалевидная мошонка, фимоз, крипторхизм, паховые грыжи, умственная отсталость.
Собственно брахидактилия сопровождается патологическим укорочением пальцев рук и ног, иногда – их сплющенной формой или расщеплением. В зависимости от тяжести патологии, брахидактилия может сочетаться с тугоподвижностью в суставах (симфалангией), синдактилией, полидактилией, мышечной слабостью, в комплексе нарушающими нормальное функционирование кисти или стопы. При брахидактилии часто наблюдается гипоплазия или аплазия ногтевых пластин.
Диагностика
Брахидактилия у ребенка может диагностироваться в пренатальном или постнатальном периоде. Дородовая диагностика может включать консультацию генетика (при наличии семейного анамнеза брахидактилии) и 3D УЗИ, проводимое в периоде ведения беременности на гестационном сроке 20-24 недели. Выявление изолированной брахидактилии не является основанием для искусственного прерывания беременности; при обнаружении хромосомных синдромов вопрос решается в индивидуальном порядке.
После рождения диагноз брахидактилии устанавливается детским травматологом-ортопедом на основании клинического обследования и рентгенологических данных (рентгенографии пальцев кисти, костей кисти, пальцев стопы, костей стопы). Решающее значение имеют результаты ДНК-диагностики.
Лечение
В лечении брахидактилии используются как консервативные, так и хирургические методы. В ряде случаев для восстановления функции кисти или стопы достаточным является проведение курсов ЛФК и массажа. Это позволяет улучшить работу мышечно-связочного аппарата и избежать ограничения двигательной активности.
Целью хирургической коррекции брахидактилии служит изменение линейных размеров кисти, устранение сопутствующей симфалангии и синдактилии. С целью коррекции линейных размеров пальцев может проводиться дистракция, поллицизация, микрохирургическая аутотрансплантация пальца стопы на кисть. Операция при синдактилии предполагает разделение сросшихся пальцев с кожной, сухожильно-мышечной или костной пластикой. Оперативное лечение брахидактилии направлено, прежде всего, на максимальное восстановление функции кисти и вторично – на достижение приемлемого эстетического результата.
Профилактика
Специфическая профилактика не разработана ввиду того, что брахидактилия является генетически обусловленной, наследуемой аномалией.
Профилактическая работа предусматривает проведение медико-генетического консультирования семей с отягощенной наследственностью для определения вероятности рождения у них ребенка с брахидактилией; исключение родственных браков.
Источник