
Атаксия пьер мари мкб код

Связанные заболевания и их лечение
Описания заболеваний
Содержание
- Описание
- Дополнительные факты
- Причины
- Течение и стадии
- Симптомы
- Диагностика
- Лечение
- Прогноз
Названия
Название: Наследственная мозжечковая атаксия Пьера-Мари.
Наследственная мозжечковая атаксия Пьера-Мари
Описание
Наследственная мозжечковая атаксия Пьера. Мари – генетически детерминированное неуклонно прогрессирующее поражение мозжечка, связанное с его дегенеративными изменениями. Развивается после 20 лет. В клинической картине мозжечковая атаксия сочетается с гиперрефлексией, офтальмологическими расстройствами и снижением интеллекта. Диагностический алгоритм предусматривает неврологический и офтальмологический осмотр, МРТ головного мозга, УЗДГ или МРА церебральных сосудов, генетическое консультирование. Радикальная терапия не разработана, осуществляется симптоматическое лечение антидепрессантами, миорелаксантами, седативными и ноотропами. Рекомендуется ЛФК, витаминотерапия и водолечение.
Дополнительные факты
Наследственная мозжечковая атаксия была подробно описана Пьером Мари в 1893г. Как отличающаяся от известной на то время атаксии Фридрейха нозологическая форма. Действительно, атаксия Пьера-Мари имеет другой тип наследования, более старший возраст манифестации и свои клинические отличия. В связи с этим, несмотря на некоторую общность морфологического субстрата этих заболеваний в виде дегенерации тканей мозжечка и его проводящих путей, в современной неврологии окончательно утвердилось их выделение в самостоятельные нозологические единицы.
Как правило, атаксия Пьера-Мари манифестирует в возрасте от 20 до 45 лет. Мужчины и женщины заболевают одинаково часто. Распространенность патологии составляет 1 случай на 200 тыс. Человек. Морфологические изменения при наследственной мозжечковой атаксии преобладают в тканях ядер и коры мозжечка, менее выражено поражение спиноцеребеллярных путей и боковых канатиков спинного мозга, процессы дегенерации могут наблюдаться в ядрах моста и продолговатого мозга.
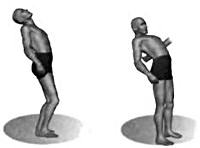
Наследственная мозжечковая атаксия Пьера-Мари
Причины
Наследственная мозжечковая атаксия имеет доминантный механизм наследования, т. Е. Развивается при получении дефектного гена от одного из родителей. Зачастую экзогенные причины выступают в роли триггеров, провоцирующих начало заболевания и усугубляющих его течение. К подобным факторам относятся: различные инфекции (брюшной тиф, сыпной тиф, бруцеллез, сальмонеллез, дизентерия, иерсиниоз, бактериальная пневмония, пиелонефрит и тд ), беременность, травмы (ЧМТ, повреждения грудной клетки, переломы таза, сильные ожоги), интоксикации.
Течение и стадии
Атаксия Пьера-Мари характеризуется постоянным и неуклонно прогрессирующим нарастанием патологической симптоматики. Выделение периодов обострения заболевания и его ремиссии не наблюдается. Под влиянием перенесенных инфекционных заболеваний и прочих экзогенных воздействий атаксия Пьера-Мари может изменить характер своего течения, что значительно затрудняет ее диагностику. Так, после перенесенной инфекции наблюдается резкое ухудшение состояния пациента с последующим частичным восстановлением, имитирующим улучшение болезни.
Симптомы
Ведущим симптомокомплексом заболевания является мозжечковая атаксия. Она включает нарушения походки с отклонением тела в стороны, расстройство статики (в позе Ромберга наблюдается падение в сторону или назад), дискоординацию движений (гиперметрию, дисдиадохокинез, промахивание при выполнении пальце-носовой пробы), размашистую макрографию, дизартрию с прерывистой и замедленной речью, интенционный тремор. Манифестация симптомов обычно происходит с легких нарушений походки, иногда первыми проявлениями становятся боли в пояснице и ногах, по описанию самих пациентов имеющие «стреляющий» характер. Затем возникает атаксия в руках, зачастую сопровождающаяся тремором. В конечностях, лице и на туловище могут наблюдаться непроизвольные мышечные подергивания. Во многих случаях наблюдается снижение мышечной силы. Чувствительность, как правило, сохранена.
Ассоциированные симптомы: Боль в пояснице. Макрография. Тремор. Тремор рук.
Диагностика
При наличии типичной клиники и прослеживании ее в нескольких поколениях диагноз не представляет для невролога особых затруднений. Спорадические случаи заболевания требуют более углубленного обследования пациента и тщательной дифдиагностики с другими видами мозжечковой атаксии, рассеянным склерозом, нейросифилисом.
При необходимости проводят исключение приобретенной органической патологии: опухолей мозжечка (медуллобластомы, астроцитомы, гемангиобластомы), церебеллита, сосудистых нарушений, сдавлений мозжечка при гематомах или абсцессах головного мозга, окклюзионной гидроцефалии. Для этого используют КТ и МРТ головного мозга, УЗДГ и МРА церебральных сосудов.
Диагностику офтальмологических расстройств проводит офтальмолог. Обследование включает проверку остроты зрения, исследование конвергенции, офтальмоскопию, периметрию, измерение угла косоглазия и пр. Для более точной верификации диагноза может потребоваться консультация генетика.
Лечение
В лечении пациентов наряду с неврологами принимают участие офтальмологи и психиатры. Поскольку этиотропная терапия пока не разработана, применяется симптоматическое лечение. В основном это антидепрессанты (амитриптилин, флуоксетин, циталопрам, тетриндол), седативные средства (валериана, пион, магния сульфат), ноотропы (гамма-аминомасляная кислота, пирацетам, экстракты гинго-билоба), препараты, уменьшающие мышечный тонус (меликтин, баклофен, кондельфин). Рекомендованы витамины гр. В, вит. РР и вит. С; бальнеотерапия, лечебная физкультура. Не маловажное значение имеет соблюдение правильного рабочего режима, исключение физических и психологических перегрузок.
Прогноз
Прогноз относительно выздоровления неблагоприятный. Симптомы заболевания постоянно усугубляются и приводят к инвалидизации. Однако систематическое применение симптоматического лечения, регулярное выполнение специального комплекса ЛФК и других рекомендаций делают благоприятным прогноз для жизни пациента.
Источник
Наследственная мозжечковая атаксия Пьера-Мари — генетико-семейное заболевание, обусловленное прогрессирующим мозжечковым расстройством, отягощенным поражением пирамидальных путей. Характеризуется повышенным рефлексом сухожилий, скандированной речью, дисбалансом координации движений, нарушением зрения и глазодвигательной моторики. Характер наследования — аутосомно-доминантный. Мутантный ген имеет высокую популяцию: пропуск поколений встречается редко.
Содержание статьи:
- Причины и течение атаксии Пьера-Мари
- Симптомы атаксии Пьера-Мари
- Дифференциальная диагностика атаксии Пьера-Мари
- Диагностика атаксии Пьера-Мари
- Лечение и прогноз атаксии Пьера-Мари

Среди наследственных болезней спиноцеребеллярные атаксии стоят по частоте заболеваемости на втором месте после нервно-мышечных патологий. По статистическим данной мозжечковой атаксией Пьера-Мари страдают 1 человек на 200 тыс. населения.
Генетическое расстройство в детском и подростковом возрасте протекает бессимптомно и проявляется начиная с третьего десятка жизни.
Причины и течение атаксии Пьера-Мари
Поражение функций мозжечка обусловлено генетической патологией по аутосомно-доминантному типу наследования. Для развития атаксии достаточно генетического нарушения, унаследованного от одного родителя.
Мозжечок — основной координаторный центр, выполняющий двигательные задачи. Полушария его отвечают за согласованность движений, а червь мозжечка — за устойчивость и равновесие.
Патологоанатомические признаки болезни выражены гипоплазией мозжечка, уменьшением нижних олив и истощением варолиева моста. На фоне этого, как правило, происходит дегенерация спиноцеребральных путей, разрушение клеток коры мозжечка и ядер, дегенеративные нарушения продолговатого мозга и в ядрах моста мозга.
В зависимости от сосредоточения мозжечкового поражения атаксию разделяют на динамическую и статико-локомоторную. В первом случае патологические нарушения обнаруживаются в полушариях, что обуславливает десинхронизацию мышечных ритмов (дисметрия, скандированная речь, непроизвольное дрожание туловища, головы, конечностей и др.) При статико-локомоторной форме затронут червь, что вызывает расстройство походки, устойчивости и равновесия.
Несмотря на врожденный характер атаксия Пьера-Мари манифестирует, начиная с 20 лет и старше. Провоцирующими факторами выступают инфекционные болезни (сальмонеллез, зоонозная инфекция, бактериальная пневмония, брюшной или сыпной тиф, пиелонефрит, менингит и пр.). В качестве экзогенных причин могут послужить черепно-мозговая травма, перелом тазовых костей или грудной клетки, глубокие ожоги и интоксикация различной природы.
Наследственная мозжечковая патология отличается непрерывно прогрессирующими проявлениями. Симптоматическая терапия не обеспечивает периодов ремиссии. Внешние патогенные факторы в виде различных болезней ухудшают состояние пациента. В дальнейшем тяжелое состояние проходит и возвращается типичный симптомокомплекс мозжечковой патологии.
Симптомы атаксии Пьера-Мари
Основным признаком наследственной болезни будут нервно-мышечные нарушения моторики, которые не ограничиваются отдельной группой мышц или конкретными движениями.
Мозжечковую атаксию отличают характерные симптомы:
- нарушение походки;
- расстройство статики;
- тремор конечностей и тела;
- мышечные подергивания;
- непроизвольные частые колебательные движения глаз;
- медленная речь;
- изменение почерка в сторону значительного увеличения букв;
- снижение мышечного тонуса.
Атаксия начинает развиваться с нарушения в походке: больной передвигается покачиваясь. Иногда первыми симптомами будут прострелы в поясничной области. Затем патология затрагивает руки, отмечается их дрожание.
При болезни Пьера-Мари можно наблюдать парез конечностей, на фоне которого повышены сухожильные рефлексы. Нередко у пациента фиксируют сгибательные и разгибательные пирамидные рефлексы стоп. Достаточно часто встречаются церебральные симптомы: опущение верхнего века (птоз), затрудненная конвергация глаз, атрофия зрительно нерва.
У 50% больных наблюдаются психические и умственные нарушения: деменция, олигофрения, депрессия.
Дифференциальная диагностика атаксии Пьера-Мари
Немаловажное значение в диагностике имеет скрупулёзный сбор сведений генетической заболеваемости ближайших родственников и особенностей клинической картины.
Диагностика предполагает проведение лабораторных и инструментальных исследований:
- Электроэнцефалография (ЭЭГ). Обнаруживает диффузную дельта/тета-активность и ослабление альфа-ритма;
- Электромиография. Выявляет аксонально-демиелинизирующее нарушение волокон периферических нервов;
- Магнитно-резонансная томография. Фиксирует морфологические изменения структур спинного и головного мозга;
- ДНК-тест. Определяет генетическую природу атаксии;
- Лабораторные анализы. Позволяют распознать нарушение обмена аминокислот.
Единичный случай в семье мозжечковой атаксии требует более глубокого обследования и дифференциальной диагностики. Помимо вышеперечисленных заболеваний, имеющих симптомокомплекс атаксии, проводят освидетельствование на предмет исключения новообразования мозжечка, абсцесса или гематомы головного мозга, церебеллита и гидроцефалии.
При офтальмологических расстройствах требуется обследование у соответствующего специалиста.
Для подтверждения предварительного диагноза семейной атаксии необходима консультация генетика.
Диагностика атаксии Пьера-Мари
Симптомокомплекс мозжечковой атаксии идентичен клинической картине наследственной атаксии Фридрейха. Поэтому при постановке диагноза возникают трудности.
Основное отличие — тип наследования. Доминантное наследование характерно для мозжечкового заболевания Пьера-Мари. Рецессивный вид свойственен атаксии Фридрейха. Учитывается возраст, в котором проявились симптомы заболевания. Более ранняя манифестация свойственна аутосомно-рецессивному характеру болезни.
Невролог исследует изменения сухожильных рефлексов, которые увеличены при мозжечковой форме атаксии и понижены при болезни Фридрейха. Кроме того, атаксии Пьера-Мари не характерны костные деформации и потеря чувствительности.
Весьма затруднительно дифференцировать рассеянный склероз и мозжечковую атаксию. Для обоих заболеваний присущи пирамидные дефекты стоп, глазодвигательные расстройства, а также нервно-мышечные нарушения моторики. Однако при рассеянном склерозе в противовес атаксии возможны периоды ремиссии. Кроме того, отличительной чертой склероза являются глубокий парапарез и более выраженные тазовые нарушения.
Лечение и прогноз атаксии Пьера-Мари
Ведущим врачом в данном случае является невропатолог. Он разрабатывает схему консервативной терапии, которая направлена на нивелирование симптоматики и включает в себя:
- Общеукрепляющий медикаментозный комплекс. Назначаются препараты, подавляющие фермент холинэстераза (дезагреганты), предупреждающие повреждение нейронов мозга (нейропротекторы), витамины группы РР, В и С;
- Лечебная физкультура, кинезиотерапия — основные реабилитационные мероприятия. Задача тренировок — лечение движением, укрепление мышц и ослабление симптома дискоординации. При статистической мозжечковой атаксии подбираются упражнения для тренировки равновесия. Для динамической атаксии разрабатывается тренировочный комплекс, повышающий согласованность и точность движений.
- Физиотерапия. Проводится с целью предупреждения контрактуры конечностей, атрофии мышц, коррекции походки, улучшения координации, поддержки общефизической формы;
- Массаж, мануальная- и рефлексотерапия. Проводится для улучшения обменных процессов.
Прогноз наследственной атаксии Пьера-Мари неблагоприятный для трудовой деятельности. Симптомы прогрессируют на протяжении жизни, трудоспособность снижается, а психические нарушения усугубляются. Больной инвалидизируется.
Тем не менее при условии постоянного выполнения симптоматической терапии и соблюдения щадящего режима, прогноз для жизни хороший.
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Течение и стадии
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: G11 Наследственная атаксия.
G11 Наследственная атаксия
Описание
Наследственная мозжечковая атаксия Пьера. Мари – генетически детерминированное неуклонно прогрессирующее поражение мозжечка, связанное с его дегенеративными изменениями. Развивается после 20 лет. В клинической картине мозжечковая атаксия сочетается с гиперрефлексией, офтальмологическими расстройствами и снижением интеллекта. Диагностический алгоритм предусматривает неврологический и офтальмологический осмотр, МРТ головного мозга, УЗДГ или МРА церебральных сосудов, генетическое консультирование. Радикальная терапия не разработана, осуществляется симптоматическое лечение антидепрессантами, миорелаксантами, седативными и ноотропами. Рекомендуется ЛФК, витаминотерапия и водолечение.
Дополнительные факты
Наследственная мозжечковая атаксия была подробно описана Пьером Мари в 1893г. Как отличающаяся от известной на то время атаксии Фридрейха нозологическая форма. Действительно, атаксия Пьера-Мари имеет другой тип наследования, более старший возраст манифестации и свои клинические отличия. В связи с этим, несмотря на некоторую общность морфологического субстрата этих заболеваний в виде дегенерации тканей мозжечка и его проводящих путей, в современной неврологии окончательно утвердилось их выделение в самостоятельные нозологические единицы.
Как правило, атаксия Пьера-Мари манифестирует в возрасте от 20 до 45 лет. Мужчины и женщины заболевают одинаково часто. Распространенность патологии составляет 1 случай на 200 тыс. Человек. Морфологические изменения при наследственной мозжечковой атаксии преобладают в тканях ядер и коры мозжечка, менее выражено поражение спиноцеребеллярных путей и боковых канатиков спинного мозга, процессы дегенерации могут наблюдаться в ядрах моста и продолговатого мозга.
G11 Наследственная атаксия
Причины
Наследственная мозжечковая атаксия имеет доминантный механизм наследования, т. Е. Развивается при получении дефектного гена от одного из родителей. Зачастую экзогенные причины выступают в роли триггеров, провоцирующих начало заболевания и усугубляющих его течение. К подобным факторам относятся: различные инфекции (брюшной тиф, сыпной тиф, бруцеллез, сальмонеллез, дизентерия, иерсиниоз, бактериальная пневмония, пиелонефрит и тд ), беременность, травмы (ЧМТ, повреждения грудной клетки, переломы таза, сильные ожоги), интоксикации.
Течение и стадии
Атаксия Пьера-Мари характеризуется постоянным и неуклонно прогрессирующим нарастанием патологической симптоматики. Выделение периодов обострения заболевания и его ремиссии не наблюдается. Под влиянием перенесенных инфекционных заболеваний и прочих экзогенных воздействий атаксия Пьера-Мари может изменить характер своего течения, что значительно затрудняет ее диагностику. Так, после перенесенной инфекции наблюдается резкое ухудшение состояния пациента с последующим частичным восстановлением, имитирующим улучшение болезни.
Симптомы
Ведущим симптомокомплексом заболевания является мозжечковая атаксия. Она включает нарушения походки с отклонением тела в стороны, расстройство статики (в позе Ромберга наблюдается падение в сторону или назад), дискоординацию движений (гиперметрию, дисдиадохокинез, промахивание при выполнении пальце-носовой пробы), размашистую макрографию, дизартрию с прерывистой и замедленной речью, интенционный тремор. Манифестация симптомов обычно происходит с легких нарушений походки, иногда первыми проявлениями становятся боли в пояснице и ногах, по описанию самих пациентов имеющие «стреляющий» характер. Затем возникает атаксия в руках, зачастую сопровождающаяся тремором. В конечностях, лице и на туловище могут наблюдаться непроизвольные мышечные подергивания. Во многих случаях наблюдается снижение мышечной силы. Чувствительность, как правило, сохранена.
Диагностика
При наличии типичной клиники и прослеживании ее в нескольких поколениях диагноз не представляет для невролога особых затруднений. Спорадические случаи заболевания требуют более углубленного обследования пациента и тщательной дифдиагностики с другими видами мозжечковой атаксии, рассеянным склерозом, нейросифилисом.
При необходимости проводят исключение приобретенной органической патологии: опухолей мозжечка (медуллобластомы, астроцитомы, гемангиобластомы), церебеллита, сосудистых нарушений, сдавлений мозжечка при гематомах или абсцессах головного мозга, окклюзионной гидроцефалии. Для этого используют КТ и МРТ головного мозга, УЗДГ и МРА церебральных сосудов.
Диагностику офтальмологических расстройств проводит офтальмолог. Обследование включает проверку остроты зрения, исследование конвергенции, офтальмоскопию, периметрию, измерение угла косоглазия и пр. Для более точной верификации диагноза может потребоваться консультация генетика.
Лечение
В лечении пациентов наряду с неврологами принимают участие офтальмологи и психиатры. Поскольку этиотропная терапия пока не разработана, применяется симптоматическое лечение. В основном это антидепрессанты (амитриптилин, флуоксетин, циталопрам, тетриндол), седативные средства (валериана, пион, магния сульфат), ноотропы (гамма-аминомасляная кислота, пирацетам, экстракты гинго-билоба), препараты, уменьшающие мышечный тонус (меликтин, баклофен, кондельфин). Рекомендованы витамины гр. В, вит. РР и вит. С; бальнеотерапия, лечебная физкультура. Не маловажное значение имеет соблюдение правильного рабочего режима, исключение физических и психологических перегрузок.
Прогноз
Прогноз относительно выздоровления неблагоприятный. Симптомы заболевания постоянно усугубляются и приводят к инвалидизации. Однако систематическое применение симптоматического лечения, регулярное выполнение специального комплекса ЛФК и других рекомендаций делают благоприятным прогноз для жизни пациента.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник