
Болезнь маделунга код по мкб

Содержание
- Синонимы диагноза
- Описание
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
Другие названия и синонимы
Диффузная липома шеи, Диффузный симметричный липоматоз, Множественный доброкачественный липоматоз, Синдром Лонуа-Бансода.
Названия
Болезнь Маделунга.
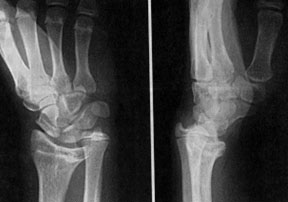
Рентгенограмма кисти с деформацией Маделунга
Синонимы диагноза
Диффузная липома шеи, Диффузный симметричный липоматоз, Множественный доброкачественный липоматоз, Синдром Лонуа-Бансода.
Описание
Маделунга болезнь (синоним: деформация Маделунга, хронический подвывих кисти)локальная физарная дисплазия, характеризующаяся укорочением лучевой кости и вывихом (подвывихом) локтевой кости, что внешне проявляется выстоянном головки локтевой кости и смещением кисти в ладонную сторону.
Симптомы
Костная деформация обусловливает своеобразную штыкообразную деформацию лучезапястного сустава, которая развивается вследствие преждевременного закрытия дистальной эпифизарной зоны роста лучевой кости, что ведет к прогрессирующему ее укорочению. Чаще поражается одна конечность. Иногда болезнь Маделунга сочетается с диспластическим сколиозом.
У девочек болезнь Маделунга встречается в 34 раза чаще, чем у мальчиков. Первые ее признаки обычно появляются в возрасте 9-12 лет, т. Е. Во время бурного роста длинных трубчатых костей. Нередко болезнь Маделунга необоснованно связывают с возможными травмами сустава, перенесенными воспалительными процессами. Вначале отмечают косметический дефект, ограничение движений кисти в тыльную и локтевую стороны. Деформация с возрастом увеличивается, появляются боли и ощущение усталости при движениях, обусловленные инконгруэнтностью суставных поверхностей.
Судороги.
Диагностика
Диагноз устанавливают на основании характерных клинических и рентгенологических проявлений. При рентгенологическом исследовании выявляют относительное укорочение лучевой кости на 45 см и скошенность суставной поверхности дистального эпифиза лучевой кости в ладонную и локтевую сторону, что создает впечатление подвывиха кисти (костей запястья). Полулунная кость как бы нависает над ладонным краем эпифиза лучевой кости. Высота дистального эпифиза по лучевой стороне значительно превосходит его высоту по локтевой стороне, а раннее закрытие эпифизарной ростковой зоны по локтевой и ладонной стороне создает ладонный и локтевой наклон суставной поверхности. Отмечают изменение расположения проксимального ряда костей запястья, они образуют клин, на вершине которого располагается полулунная кость. Головка локтевой кости находится в положении вывиха (в дистальном направлении) и смещена в тыльном направлении относительно лучезапястного сустава. Обычно расположена на уровне основания пястных костей.
Дифференциальная диагностика
Болезнь Маделунга следует дифференцировать с посттравматической деформацией типа Маделунга (например, в результате эпифизеолиза), лучевой косорукостью, штыковидными деформациями после перенесенного остеомиелита, при опухолях и Эта болезнь напоминает мезомелическую дисплазию, или синдром Лери-Вейлля, который относится к дисхондростеозам. Для синдрома Лери-Вейлля характерны двустороннее поражение, малый рост больного, короткая кисть. Иногда он сопровождается деформациями большеберцовой и плечевой костей. Обе лучевые кости при этом заболевании изогнуты под углом, открытым в локтевую сторону, в результате чего суставные поверхности дистальных эпифизов лучевых костей скошены в локтевую сторону. Искривление более выражено на уровне дистальной трети кости. Отмечается вывих головок локтевых костей в проксимальном направлении из-за относительного укорочения этих костей. Кисть отклонена в локтевую сторону.
Лечение
Консервативное лечение болезни Маделунга неэффективно. Операцию следует выполнять в возрасте 1314 лет. Основными показаниями к ней являются выраженный косметический дефект, нарушение функции и боли в суставе. Более раннее хирургическое вмешательство, направленное на устранение косметического дефекта, например резекция головки локтевой в сочетании с корригирующей остеотомией лучевой кости, часто приводит к значительному нарушению функции лучезапястного сустава. Остеотомия укороченной лучевой кости на уровне ее дистального метафиза и применение дистракционно-компрессионных аппаратов позволяет одновременно добиться удлинения кости за счет образования костного регенерата и смещения дистального фрагмента лучевой кости в тыльную и лучевую сторону, что обеспечивает восстановление конгруэнтности, ликвидацию подвывиха костей запястья и вывиха головки локтевой кости.
Прогноз
Прогноз в отношении функций после оперативного лечения благоприятный. При нерациональном лечении развивается остеоартроз лучезапястного сустава и сохраняется косметический дефект.
Источник
Болезнь Маделунга – это заболевание неясной этиологии, которое характеризуется прогрессирующим разрастанием жировой клетчатки в области шеи. Множественные, симметрично расположенные липомы развиваются на плечах, передне-боковых и задней поверхностях шеи, верхней части спины. В области ключиц, в проекции дельтовидных мышц и по верхней трети спины формируются выраженные жировые подушки. Постепенно нарастает тугоподвижность шеи, затрудняется глотание и дыхание. Диагноз ставится на основании клинической картины болезни и данных биопсии. Хирургическое лечение проводится с косметической целью и для устранения симптомов сдавления жизненно важных органов.
Общие сведения
Болезнь Маделунга – множественный доброкачественный липоматоз, диффузная липома шеи, диффузный симметричный липоматоз, синдром Лонуа-Бансода. Первые описания случаев заболевания в научной литературе датируются 1848 годом. Спустя 40 лет известный немецкий хирург Отто фон Маделунг опубликовал обзор 33 случаев заболевания. Доброкачественный липоматоз относится к числу редко встречающейся патологии. Он диагностируется у мужчин и женщин старше 30 лет. Соотношение пациентов мужского и женского пола составляет 15:1 – 30:1. У большинства больных имеется алкогольный анамнез.

Болезнь Маделунга
Причины
В настоящее время описано порядка 300 случаев болезни Маделунга, что явно недостаточно для того, чтобы достоверно установить механизм развития патологии. Большинство наблюдений не содержит сведений, позволяющих судить об условиях, в которых манифестировало заболевание. Рассматриваются четыре наиболее вероятные причины нарушения обмена липидов у больных доброкачественным липоматозом:
- Генетическая предрасположенность. Преобладающий тип наследования ‒ аутосомно-доминантный. При нем носитель дефектного гена страдает патологией сам и передает ее половине своих потомков. Мальчики и девочки заболевают одинаково часто. Описаны семейные случаи болезни Маделунга, обусловленные мутациями митохондриальной ДНК, при которых заболевание передается по женской линии детям обоих полов.
- Дисфункция нервных центров. В генезе диффузного симметричного липоматоза лежит нарушение нейро-трофических влияний на жировую ткань. Поражение нервной системы в результате травмы или интоксикации приводит к нарушению баланса гормонов желез внутренней секреции, что сказывается на обмене липидов в организме неблагоприятным образом.
- Заболевание шейных лимфоузлов. Ряд исследователей считает, что появление липом является следствием аденолипоза. Поражение лимфатических узлов протекает с постепенным замещением лимфоидной ткани на жировую. Аденолипоз затрагивает преимущественно поверхностно расположенные лимфоузлы и развивается симметрично по обеим сторонам тела.
- Хронический алкоголизм. Болезнь Маделунга диагностируется преимущественно у мужчин, злоупотребляющих спиртными напитками. Описаны случаи выявления патологии у детей и непьющих женщин, но число этих случаев ничтожно мало. Алкоголь, по мнению ряда исследователей, становится тем «пусковым» фактором, который позволяет реализоваться имеющейся у пациента генетической предрасположенности.
К числу предрасполагающих факторов развития симметричного липоматоза можно отнести наличие избыточного веса, гиподинамию, преобладание в рационе жиров при выраженном дефиците белка, употребление пищи преимущественно в вечерние и ночные часы. Активному отложению ацетилглицеролов в жировых депо способствует недостаточно высокий уровень гормона щитовидной железы трийодтиронина, разрастание бурой жировой ткани.
Патогенез
Существуют две основные теории развития болезни Маделунга. Согласно первой, в области липом идет накопление эмбрионального бурого жира. Вторая предполагает наличие дефекта адренергически стимулированного липолиза. При этом на поверхности стволовых жировых клеток липом начинают экспрессироваться по аналогии с бурой жировой тканью разобщающие белки-1. Концентрация этих белков не увеличивается в ответ на воздействие катехоламинов, несмотря на наличие в клеточной мембране адренорецепторов.
Нарастание концентрации катехоламинов не влияет на процесс распада жира в клетках липом, не стимулирует экспрессию фермента синтазы оксида азота. Вместе с тем, донаторы оксида азота ингибируют деление жировых клеток, формирующих липому, избирательно подавляют пролиферацию, стимулированную синтазой оксида азота.
Классификация
Все случаи диагностированной болезни Маделунга можно разделить по признаку локального и системного поражения. Однако нельзя исключать вероятность того, что, начавшись как местный процесс, заболевание не приобретет системный характер под влиянием провоцирующих факторов. Учитывая расположение липом на теле, выделяют три основных типа болезни:
- Тип I. Встречается в подавляющем большинстве случаев (порядка 70%). Липомы располагаются в затылочной и шейной области, образуя изолированное утолщение в форме «хомута».
- Тип II. Системное поражение, при котором липомы располагаются симметрично на всей поверхности тела. Поражается плечевой пояс, грудная клетка, бедра, живот. В ряде случаев расположение гипертрофированных жировых отложений визуально создает псевдоатлетический силуэт (это так называемый псевдоатлетический синдром).
- Тип III. Редко встречающийся гинекоидный тип, при котором большинство липом формируется в области бедер.
Симптомы болезни Маделунга
Первые признаки патологии у разных пациентов могут существенно отличаться. В большинстве случаев дебют заболевания связывают с разрастанием жировой прослойки в области затылка или же одновременно затылка и переднее-боковых областей шеи. Жировая подушка спускается по направлению к 7-му шейному позвонку, заполняет надключичные области, распространяется на верхнюю часть спины, формируя заметный «бычий» горб. Кольцевидная липома растет медленно, но с годами может достигать толщины 15 см и веса 6-8 кг.
Второй часто встречающийся вариант – развитие множественных липом по сторонам шеи или по всему туловищу. Липомы имеют мягко-эластичную консистенцию, нечеткие за счет отсутствия соединительнотканной капсулы контуры. Жировики легко смещаются относительно покрывающей их неизмененной кожи и подлежащих тканей. Появившись однажды, такие уплотнения могут существовать десятилетиями, пока под воздействием провоцирующих факторов не переходят в активный рост. Узлы с фиброзной капсулой могут формироваться на боковых поверхностях шеи по мере роста кольцевидной липомы.
Сначала узлы безболезненные, пальпация не вызывает у пациента неприятных ощущений. Затем может присоединиться чувство напряжения в основании черепа. Сдавление органов шеи и средостения проявляется целым комплексом симптомов. Больные предъявляют жалобы на головные боли, боли в области сердца, сердцебиение, одышку, затруднение дыхания, нарушение глотания и артикуляции. Размер узлов не меняется при общем снижении веса. При развитии некроза в толще липомы образование спадается. При гнойном расплавлении содержимого локальное уплотнение начинает флюктуировать.
На фоне болезни Маделунга могут появиться сенсорная, моторная и вегетативная дистонии, судорожные припадки, псевдомиопатия, сопровождающаяся нарастающей мышечной слабостью. Пациенты с поражением мышц не могут поднять руки выше определенного уровня, причесаться, заложить руки за голову, выполнить другие простые действия. У женщин часто выявляется олигоменорея.
Осложнения
Накопление жировой ткани затрудняет движения, деформирует контуры тела человека, создавая выраженное эстетическое несовершенство. Угрожающими для жизни последствиями болезни Маделунга становятся сдавление жировой тканью пищевода, трахеи и крупных бронхов, сонной артерии, ветвей верхней полой вены. Описаны случаи злокачественного перерождения изначально доброкачественных разрастаний жировой ткани.
Согласно анализу причин смертности пациентов с липоматозом, преобладает так называемая «внезапная смерть», когда больной в удовлетворительном состоянии умирает без видимой на то причины. Такая внезапная смерть может быть следствием имеющегося у человека алкогольного анамнеза или же хронического сдавления органов средостения.
Диагностика
Обследованием пациентов с болезнью Маделунга занимается врач-дерматолог. В случае необходимости к диагностике могут подключаться другие узкие специалисты: эндокринолог, хирург. Пациенту назначается комплексное всестороннее обследование с целью определения основного и сопутствующих заболеваний, оценки общего состояния здоровья, выявления возможных противопоказаний к проведению оперативного вмешательства. План обследования включает:
- Анализы крови и мочи. Характерным для больных синдромом Лонуа-Бансода является гиперальфапротеинемия на фоне повышенной активности липопротеиновой липазы. Также могут выявляться повышенные или, наоборот, сниженные уровни гормонов надпочечников, щитовидной и других желез, нарушения в работе печени, почек и других внутренних органов.
- Гистологическое исследование. Образцы тканей, взятые из липом методом биопсии, исследуются под микроскопом. По морфологическому строению узлы, развивающиеся при болезни Маделунга, состоят из неизмененной жировой ткани. На этих данных во многом основывается дифференциальная диагностика доброкачественного липоматоза с другими заболеваниями.
- Методы медицинской визуализации. Получить необходимые сведения о характере разрастаний позволяют УЗИ мягких тканей, МРТ области головы и шеи, КТ области средостения. Наиболее предпочтительный и информативный метод исследования определяет врач на основании картины заболевания. Все методы позволяют оценить состояние органов средостения и шеи, оценить размеры жировых узлов, их доброкачественный характер.
Дифференциальная диагностика симметричного липоматоза проводится с лимфосаркомой, болезнью Деркума, нейрофиброматозом. На первый план в сборе анамнеза выходит уточнение перечня лекарств, которые пациент принимает курсами или же постоянно, для исключения индуцированного лекарствами липоматоза.
Лечение болезни Маделунга
Операция – это единственный способ удалить жировые разрастания на теле пациента. Липомы не чувствительны к фармакологическим, физиотерапевтическим и другим воздействиям. В зависимости от целей, стоящих перед пациентом и его лечащим врачом, наиболее предпочтительным может быть один из следующих методов:
- Липосакция. Аспирация жира у пациентов с болезнью Маделунга проводится преимущественно по эстетическим показаниям. Выбор наиболее предпочтительного способа липосакции определяется объемом липом и жировых подушек. Механическая липоаспирация позволяет убрать небольшие по объему наплывы жира. При значительных по размеру образованиях предпочтительными являются ротационная и ультразвуковая липосакция.
- Хирургическая операция. Иссечение липом – метод выбора в тех случаях, когда необходимо освободить органы от сдавливающих их напластований жира. Для этого необходимо проводить все манипуляции с ювелирной тонностью, что невозможно сделать при липоаспирации канюлей вслепую. Объем операции определяется в индивидуальном порядке с учетом состояния пациента и преобладающих симптомов.
Осложняет проведение оперативных вмешательств у больных синдромом Маделунга тот факт, что липомы растут преимущественно из межфасциальной жировой ткани. Эта ткань в виде тонких прослоек проникает в межмышечные пространства. Хирург при удалении кольцевидной липомы встречается с многочисленными техническими трудностями, особенно при работе на переднебоковой поверхности шеи.
Прогноз и профилактика
Течение болезни Маделунга хроническое. Длительные периоды относительно стабильного существования липом могут сменяться их бурным ростом. Консервативное лечение неэффективно. После оперативного удаления жировых разрастаний часто возникают рецидивы, связанные с глубоким проникновением измененной жировой ткани в промежутки между мышцами и массивы нормальной подкожной жировой клетчатки.
Профилактика заболевания не разработана. С учетом того, что наследственная предрасположенность чаще всего реализуется под воздействием хронической алкогольной интоксикации, рекомендовано сократить потребление спиртных напитков или же отказаться от их употребления полностью.
Источник
Рубрика МКБ-10: E88.8
МКБ-10 / E00-E90 КЛАСС IV Болезни эндокринной системы, расстройства питания и нарушения обмена веществ / E70-E90 Нарушения обмена веществ / E88 Другие нарушения обмена веществ
Определение и общие сведения[править]
Болезнь Маделунга
Синонимы: множественный симметричный липоматоз, цефалотракальная липодистрофия,
семейный доброкачественный шейный липоматоз, липоматоз Launois-Bensaude
Болезнь Маделунга является редким заболеванием подкожных тканей, характеризуется ростом симметричных неинкапсулированных масс жировой ткани преимущественно в области лица и шеи с разнообразными клиническими проявлениями, например, снижение подвижности шеи, обструкция дыхательных путей и пр.
Болезнь Маделунга в первую очередь поражает мужчин, соотношение полов 15:1-30:1, в зависимости от региона. Болезнь Маделунга встречается во всех этнических группах.
Распространенность и заболеваемость не известны, распространенности среди мужчин в Италии оценивается порядка 1/25000. Более 300 случаев заболевания было описано на сегодняшний день.
Этиология и патогенез[править]
Патогенез болезни Маделунга до конца не изучен, но недавние исследования предполагают, что множественный симметричный липоматоз может быть связан с неполноценной норадренергической регуляцией митохондрий в тканях бурого жира. Заболевание тесно связано со злоупотреблением алкоголем (до 90% больных).
Клинические проявления[править]
Болезнь Маделунга манифестирует, как правило, в середине-конце зрелого возраста (30-60 лет). Течение заболевания характеризуется быстрым ранним развитием, с последующим медленным прогрессирующим ростом жировых отложений на лице и вокруг лица, на шее, затылке, в надключичной ямке и плечевом поясе. Также могут быть затронуты бедра. Пациенты часто изначально обращаются за медицинской помощью по эстетическим причинам и/или из-за проблем при одевании. Со временем мышцы шеи могут вызывать компрессию трахеи и пищевода, приводя к обструкции дыхательных путей и обструктивному апноэ во сне. Пациенты также часто имеют полинейропатии, заболевания печени и метаболический синдром с аномальной толерантностью к глюкозе и гиперлипидемией. Большинство пациентов страдают ожирением, но в некоторых редких случаях (10%) отмечается нормальный вес тела. В очень редких случаях описана злокачественная трансформация поражений в липосаркому .
Другие уточненные нарушения обмена веществ: Диагностика[править]
Множественный симметричный липоматоз часто ошибочно диагностируется как ожирение. Клинические признаки, демонстрирующие специфическое распределение жировой ткани являются диагностическим критерием, в частности, незатронутые дистальные отделы рук и ног. Злоупотребление алкоголем также может наводит на мысль о болезни Маделунга. Компьютерная томография или магнитно-резонансная томография могут быть использованы, чтобы подтвердить наличие симметричных, неинкапсулированных жировых отложений. Биопсия жировых масс может также использоваться, чтобы исключить липосаркому.
Дифференциальный диагноз[править]
Ожирение и липосаркома являются основными дифференциальными диагнозами болезни Маделунга. Другие нарушения, которые необходимо учитывать, включают синдром Кушинга, семейный ангиолипоматоз, синдром сливообразного живота и лимфому.
Другие уточненные нарушения обмена веществ: Лечение[править]
Некоторым пациентам лечение может не потребоваться. Лечение проводят из эстетических соображений, из-за апноэ во сне, при обструкции пищеварительного тракта, сильном болевом синдроме. Как правило, терапия болезни Маделунга включает в себя липэктомию, липосакцию (или ультразвуковую липосакцию). Прекращение употребления алкоголя и потеря веса может в некоторых случаях остановить прогрессирование заболевания.
Прогноз
Болезнь Маделунга в основном доброкачественное состояние, но в некоторых случаях может иметь серьезные физические, косметические и психологические последствия. Сопутствующие заболевания могут повлиять на прогноз. Алкоголизм играет б’ольшую роль в морбидности и смертности, чем сам множественный симметричный липоматоз.
Профилактика[править]
Прочее[править]
Мевалоновая ацидурия
Синоним: полный дефицит мевалонаткиназы
Мевалоновая ацидурия является редкой, очень тяжелой формой дефицита мевалонаткиназы, характеризуется дисморфическими особенностями, снижением прибавки массы тела, задержкой психомоторного развития, поражением глаз, гипотонией, прогрессивной атаксией, миопатией и эпизодами рецидивирующих воспалений.
Распространенность <1/1 000 000. Наследование аутосомно-рецессивное.
Расстройства окислительного фосфорилирования
Популяционная частота данной группы болезней составляет 1:10 000 живорождённых, а болезней, обусловленных дефектом митохондриальной ДНК, ориентировочно 1:8000.
Этиология
Митохондриальные заболевания, обусловленные дефектами электронного транспорта и окислительного фосфорилирования, отличаются генетической гетерогенностью, что обусловлено двойственностью генетического контроля (ядерной и митохондриальной ДНК) процессов электронного транспорта. Подавляющее большинство состояний, обусловленных ядерными мутациями, в родословной наследуются по аутосомно-рецессивному типу за исключением трихополидистрофии Менкеса.
Те заболевания, которые обусловлены мутациями митохондриальной ДНК, наследуются по материнской линии (цитоплазматическое наследование). Делеции её, как правило, в родословной встречаются спорадически. Нарушения межгеномного взаимодействия – ядерно-кодируемые множественные митохондриальные мутации и деплеции (снижение числа копий ДНК) – могут иметь аутосомно-доминантный или аутосомный тип наследственной передачи.
Патогенез
В данной группы болезней основная роль принадлежит генетически обусловленному дефициту ферментных комплексов дыхательной цепи, окислительного фосфорилирования, а также дефекту структурных митохондриальных белков и расстройствам трансмембранного транспорта специфических протеинов. В итоге происходит нарушение функционирования всей системы тканевого дыхания, страдают окислительно-восстановительные процессы в клетках, а в митохондриях и цитоплазме накапливаются недоокисленные продукты и развивается лактат-ацидоз.
Клиническая картина
Характерная особенность заболеваний, связанных с дефектом дыхательной цепи и окислительного фосфорилирования, – их прогрессирующее течение и широкий возрастной диапазон манифестации клинических симптомов – от периода новорождённости до взрослого состояния. На первый план в развёрнутой стадии болезни выступают следующие симптомы: респираторный и нейродистресс-синдром, задержка психомоторного развития, судороги, атаксия, офтальмоплегия, снижение толерантности к физической нагрузке, миопатический синдром. Кроме этого часто присоединяются признаки поражения других органов и систем: сердечно-сосудистой, эндокринной, органов зрения и слуха, почек.
У больных часто наблюдают нарушение физического и полового развития.
Диагностика
При лабораторном исследовании обнаруживают признаки, которые свойственны митохондриальным заболеваниям, – метаболический ацидоз, увеличение в крови уровней молочной и пировиноградной кислот, кетонемию, часто выявляемую только после нагрузки углеводами, снижение уровня общего карнитина, повышение экскреции с мочой органических кислот (молочной, дикарбоновых кислот, 3-метилглутаконовой, трикарбоновых кислот цикла Кребса и др.). Иногда отмечают увеличение содержания аммиака в крови и гипогликемию. В лейкоцитах или фибробластах определяют снижение активности ферментных комплексов дыхательной цепи.
В биоптатах мышечной ткани при световой микроскопии выявляют характерный феномен RRF и гистохимические признаки митохондриальной недостаточности (сниженные показатели активности ферментов дыхательной цепи). При электронной микроскопии часто обнаруживают аномальные митохондрии и изменение их количества.
Абсолютный критерий поражения мтДНК – обнаружение мутаций митохондриальной ДНК (точковые мутации, единичные и множественные делеции, дупликации и др.), которые можно выявить с помощью современных методов молекулярно-генетического анализа в биоптатах мышечной ткани. Однако отсутствие митохондриальной мутации не исключает полностью диагноз митохондриальной болезни, так как это может быть связано с наличием у больных редких мутаций, мозаичностью поражения клеток и тканей, а также возможностью повреждения ядерной ДНК.
Лечение
Лечение детей, страдающих митохондриальными заболеваниями, обусловленными дефектами электронного транспорта и окислительного фосфорилирования, должно быть поликомпонентным с назначением адекватной диеты и разнообразных лекарственных средств. Сочетанное применение препаратов, дифференцированно влияющих на разные этапы энергетического метаболизма, оказывает положительный эффект по сравнению с монотерапией отдельными медикаментами.
Особенность диетотерапии – уменьшение содержания углеводов в пищевом рационе до 10 г/кг, поскольку высокое потребление легкоусваиваемых углеводов при нарушении функции дыхательной цепи углубляет существующий дефект клеточного энергообмена.
Для коррекции процессов нарушенного транспорта электронов назначаются коэнзим Q-10 (90-200 мг/сут не менее 6 мес), янтарная кислота (5 мг/кг в сут, прерывистыми курсами по 3-4 дня и общей продолжительностью 3 мес) и цитохром С (4 мл в/м или в/в ежедневно, 3-4 курса по 10 инъекций в год).
Корректоры транспорта электронов сочетают с кофакторной терапией, улучшающей протекание энзимных реакций клеточного энергообмена (никотинамид 60-100 мг/сут, витамины В1, В2, В6 10-20 мг/сут, биотин 1-5 мг/сут), тиоктовая кислота 50-100 мг/сут, препараты левокарнитина 25-30 мг/кг в сут).
Болезнь хранения глутамил рибозо-5-фосфата
Williams et al. (1984) сообщили о случае 6-летнего мальчика с анамнезом судорожных припадков, прогрессирующим неврологическим ухудшением и протеинурией. При физикальном обследовании – умеренно грубые черты лица, генерализованная гипотония и атрофия зрительного нерва. Пациент умер от почечной недостаточности в возрасте 8 лет. Х-сцепленное рецессивное наследование было предложено исходя из семейной истории подобного расстройства у дяди по матери, у которого были судорожные припадки в возрасте 9 месяцев, с последующим ухудшением речи и зрения, а также гипертония, нефротический синдром, атрофия зрительного нерва, гипорефлексия и выраженная задержка развития, смерть натсупила от почечной недостаточности в возрасте 7 лет.
Соединение, идентифицированное как глутамил-рибозо-5-фосфат было выделено из тканей мозга и почек. Это соединение является связующей группой в АТФ-рибозилировании белков, посттрансляционная модификация, которая является важным регуляторным процессом в экспрессии генов и репарации ДНК.
Williams et al. (1984) классифицировали расстройство как гликопротеиноз. Предполагается наличие дефицита гидролазы АТФ-рибозы.
Источники (ссылки)[править]
https://www.orpha.net
Педиатрия [Электронный ресурс] / Под ред. А.А. Баранова – М. : ГЭОТАР-Медиа, 2009. – https://www.rosmedlib.ru/book/ISBN9785970410851.html
https://www.omim.org
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
- Канакинумаб
- Эзетимиб
Источник