
Болезни дауна синдромов эдвардса тернера патау

Трисомии, синдром (болезнь) Дауна, Патау, Эдвардса у ребенка, детей
Наиболее часто у человека встречаются трисомии по 21-й, 13-й и 18-й паре хромосом.
Синдром (болезнь) Дауна у детей, ребенка
Синдром (болезнь) Дауна — синдром трисомии 21 — самая частая форма хромосомной патологии у человека (1:750). У мальчиков и девочек патология встречается одинаково часто.
Достоверно установлено, что дети с синдромом Дауна чаще рождаются у пожилых родителей. Если возраст матери 35-46 лет, то вероятность рождения больного ребенка возрастает до 4,1 %. Возможность возникновения повторного случая заболевания в семье с трисомией хромосомы 21 составляет 1-2 % (с возрастом матери риск увеличивается).
Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб, широкое, плоское лицо. Типичны эпикант, запавшая спинка носа, косой (монголоидный) разрез глазных щелей, пятна Брушфильда (светлые пятна на радужке), толстые губы, утолщенный язык с глубокими бороздами, выступающий изо рта, маленькие, округлой формы, низко расположенные ушные раковины со свисающим завитком, недоразвитая верхняя челюсть, высокое нёбо, неправильный рост зубов, короткая шея.
Из пороков внутренних органов наиболее типичны пороки сердца (дефекты межжелудочковой или межпредсердной перегородок, фиброэластоз и др.) и органов пищеварения (атрезия двенадцатиперстной кишки, болезнь Гиршпрунга и др.). Среди больных с синдромом Дауна с более высокой частотой, чем в популяции, встречаются случаи лейкемии и гипотиреоза. У маленьких детей резко выражена мышечная гипотония, а у детей старшего возраста часто обнаруживается катаракта. С самого раннего возраста отмечается отставание в умственном развитии. Средняя продолжительность жизни при синдроме Дауна составляет 36 лет.
Синдром Патау у детей, ребенка
Синдром Патау — синдром трисомии 13 — встречается с частотой 1:6000. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае болезни Дауна.
При синдроме Патау наблюдаются тяжелые врожденные пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы. У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезенки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Характерна задержка умственного развития.
Большинство больных детей с синдромом Патау (98 %) умирают в возрасте до года, оставшиеся в живых страдают глубокой идиотией.
Синдром Эдвардса у детей, ребенка
Синдром Эдвардса — синдром трисомии 18 — встречается с частотой примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребенка составляет 0,7 %. У девочек встречается значительно чаще, чем у мальчиков.
Дети с трисомией 18 рождаются с низким весом, хотя сроки беременности нормальные или даже превышают норму. Фенотипические проявления синдрома Эдвардса многообразны. Наиболее часто отмечаются аномалии мозгового и лицевого черепа, мозговой череп долихоцефалической формы. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщен и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и легочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса.
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 мес, до года доживает лишь один ребенок из десяти; оставшиеся в живых — глубокие олигофрены.
Источник
Синдром Дауна
Рассмотрим некоторые хромосомные болезни. Синдром Дауна, трисомия по 21-й хромосоме – самая частая и наиболее хорошо изученная хромосомная болезнь. Частота рождения детей с синдромом Дауна составляет примерно 1:750 и не имеет какой-либо временной, этнической или географической разницы и родителей одинакового возраста. С возрастом (в большей степени матери и в меньшей мере отца) вероятность рождения ребенка с данной патологией существенно возрастает, и в возрасте 45 лет составляет около 3 %. Цитогенетические варианты синдрома Дауна разнообразны. Основную долю составляют случаи полной трисомии 21 как следствие нерасхождения хромосом в мейозе. Наряду с этим известны случаи регулярной трисомии, связанной с транслокацией 21-й хромосомы на другую – 21, 22, 13, 14 или 15-ю хромосому. Почти 50 % транслокационных форм наследуется от родителей носителей и 50 % – вновь возникшие мутации. Соотношение мальчиков и девочек среди новорожденных с синдромом Дауна составляет 1:1. Клиническая картина синдрома Дауна разнообразна: врожденные пороки развития, нарушения постнатального развития нервной системы, иммунодефициты и другие отклонения. Многие симптомы заметны уже при рождении ребенка и дальнейшем проявляются еще более отчетливо. Из черепно-лицевых дизморфий отмечается монголоидный разрез глаз, круглое уплощенное лицо, плоская спинка носа, крупный язык, брахицефалия, деформированные ушные раковины. Так же характерны мышечная гипотония и разболтанность суставов. Часто диагностируются врожденный порок сердца, клинодактилия. Встречаются изменения дерматоглифики в виде четырехпальцевой, или “обезьяньей”, складки на ладони, две кожные складки вместо трех на мизинце. Характерен низкий рост (на 20 см ниже среднего). Диагноз синдрома Дауна ставится на основании клинически на основании сочетания ряда симптомов. Наиболее важные из которых: уплощение профиля лица (90 %), отсутствие сосательного рефлекса (85 %), избыток кожи на шее (80 %), монголоидный разрез глаз (80 %), мышечная гипотония (80 %), разболтанность суставов (80 %), диспластический таз (70 %), деформированные ушные раковины (40 %), клинодактилия мизинца (60 %), четырехпальцевая сгибательная складка (поперечная линия) на ладони (40 %). Большое значение для диагностики имеет задержка умственного и физического развития ребенка. Задержка умственного развития может достигать степени имбицильности, а коэффициент умственного развития у разных детей широко варьируется (IQ от 25 до 75). Больные с синдромом Дауна часто болеют пневмониями, тяжело переносят детские инфекции. У них отмечается недостаток массы тела. Врожденные пороки внутренних органов и недостаточность иммунной системы часто приводят к летальному исходу в первые 5 лет жизни. Дифференциальная диагностика проводится с другими формами хромосомных аномалий и врожденным гипотиреозом. Цитогенетическое исследование показано и при подозрении на синдром Дауна и при клинически установленном диагнозе. В последнем случае это необходимо для прогноза здоровья будущих детей у родителей ребенка и их родственников. Лечебная помощь детям с синдромом Дауна многопланова и неспецифична. Врожденные пороки сердца устраняют оперативно. Постоянно проводится общеукрепляющая терапия, защита от действия вредных факторов внешней среды. Многие больные с трисомией 21 способны вести самостоятельную жизнь, овладевают несложными профессиями, создают семью.
Синдром Патау – трисомия по 13-й хромосоме, выделен в самостоятельную нозологическую форму в 1960 г. в результате генетического исследования у детей с врожденными пороками развития. Обнаружены простые и транслокационные формы трисомии 13, однако клинически и патологоанатомически они неразличимы. Частота синдрома Патау среди новорожденных составляет 1:6000. Соотношение полов при данной патологии близко 1:1. Частое осложнение при вынашивании плода с синдромом Патау – многоводие (50 %). Для заболевания характерны множественные, тяжелые пороки развития головного мозга, мозговой и лицевой частей черепа, внутренних органов. Окружность черепа обычно уменьшена, лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположены и деформированы (80 %). Типичный признак – расщелина верхней губы и неба (70 %). Всегда обнаруживаются пороки внутренних органов в разных комбинациях: пороки сердца (80 %), незавершенный поворот кишечника (40 %), кисты почек (42 %), аномалии внутренних половых органов (73 %), дефекты поджелудочной железы (43 %). Часто наблюдается полидактилия кистей (50 %) и их флексорное положение(44 %). Дети с синдромом Патау практически всегда имеют глубокую идиотию. Клиническая диагностика основывается на сочетании характерных пороков развития. Однако решающий фактор в диагностике – исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших больных, с целью составления прогноза для будущих детей в семье. Лечебные мероприятия неспецифичны: общеукрепляющее лечение, тщательный уход, профилактика простудных и инфекционных болезней. В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы жизни, но некоторые больные живут до нескольких лет.
Синдром Клайнфельтера относится к группе полисомий по половым хромосомам. Заболевание включает в себя случаи полисомии, при которых имеется не менее двух Х-хромосом и не менее одной Y-хромосомы. Наиболее часто (примерно 1:600) встречается синдром Клайнфельтера с набором 47,XXY.Этот синдром является и наиболее типичным клинически. Варианты полисомии с большим числом Х – и Y-хромосом (XXXY,XYY,XXXXY,XXYY) встречаются редко. Присутствие Y-хромосомы определяет формирование мужского пола. До периода полового созревания мальчики развиваются почти нормально. Вызываемый добавочной Х-хромосомой генетический дисбаланс проявляется клинически в период полового созревания в виде недоразвития семенников и вторичных мужских половых признаков. Мужчины с синдромом Клайнфельтера обычно имеют высокий рост, астеническое или евнухоидное телосложение, слабое оволосение лица, подмышечных впадин и лобка. Выявляется умственная отсталость легкой и средней степени, а в четверти случаев гинекомастия. Больные бесплодны (азооспермия, олигоспермия).
Синдром Шерешевского-Тернера – единственная форма моносомии у живорожденных. Цитогенетика синдрома разнообразна. Более половины всех больных данным синдромом имеют простую полную моносомию по Х-хромосоме (45,Х). В остальных случаях наблюдаются мозаичные формы и более редкие формы со структурными аномалиями Х-хромосом (делеция, транслокация и другие аномалии). Клинически синдром Шерешевского-Тернера проявляется следующими признаками. Со стороны половой системы отмечается либо полное отсутствие гонад (агенезия), либо гипоплазия матки и маточных труб, первичная аменорея, недостаток эстрогенов, половой инфантилизм. Встречаются различные пороки сердечно-сосудистой системы и почек. Снижения интеллекта не отмечается, однако больные обнаруживают эмоциональную неустойчивость и инфантилизм психических процессов. Внешний вид больных своеобразен. Отмечаются характерные симптомы: короткая шея с избытком кожи и крыловидными складками; в подростковом возрасте выявляется отставание в росте и развитии вторичных половых признаков; для взрослых характерны нарушения скелета, черепно-лицевые дизморфии, вальгусная девиация коленных и локтевых суставов, низкое расположение ушных раковин, диспропорции тела (укорочение ног, относительно широкий плечевой пояс, узкий таз). Рост взрослых больных на 20-30 см ниже среднего. Лечение больных с синдромом Шерешевского-Тернера комплексное и включает в себя реконструктивную и пластическую хирургию, гормональную терапию (эстрогены, гормон роста), психотерапию.
Синдром кошачьего крика – частичная моносомия по короткому плечу 5-й хромосомы (5p-). Синдром обусловлен делецией короткого плеча 5-й хромосомы. У детей с этой хромосомной аномалией отмечается необычный плач, напоминающий требовательное кошачье мяуканье или крик. Частота синдрома достаточно велика для делеционных синдромов – 1:45000. Цитогенетически в большинстве случаев выявляется делеция с утратой от трети до половины короткого плеча 5-й хромосомы, реже наблюдается полная утрата короткого плеча. Для развития клинической картины синдрома имеет значение не величина утраченного участка, а конкретный незначительный фрагмент хромосомы. Клиническая картина синдрома довольно сильно варьируется у отдельных больных по сочетанию врожденных пороков развития органов. Наиболее характерный признак – “кошачий крик” – обусловлен изменением гортани. У большинства больных имеются те или иные изменения мозгового черепа и лица: лунообразное лицо, микроцефалия, микрогения, антимонголоидный разрез глаз, высокое небо, плоская спинка носа, деформация ушных раковин. Кроме того, встречаются врожденные пороки сердца, костно-мышечной системы и внутренних органов. Выраженность клинической симптоматики меняется с возрастом. “Кошачий крик”, мышечная гипотония, лунообразность лица с возрастом исчезают, а микроцефалия выявляется более отчетливо, прогрессирует психомоторное недоразвитие, косоглазие. Продолжительность жизни больных зависит от выраженности клинической картины в целом, тяжести врожденных пороков внутренних органов (прежде всего сердца), уровня оказываемой медицинской помощи и повседневной жизни. Большинство больных умирает в первое десятилетие жизни. Во всех случаях больным и их родителям показано цитогенетическое обследование.
Источник
Синдром Дауна или трисомия по 21 хромосоме является самой частой хромосомной патологией. Из других трисомий встречаются также трисомии по 13 и 18 хромосомам.
Что такое анеуплоидия, трисомия, транслокация, мозаицизм
В каждой клетке человеческого организма находится 46 хромосом, в которых выделяют две группы: 22 пары аутосом (пронумерованных с 1 по 22, в зависимости от размера) и пара половых хромосом (XX у женщин, XY у мужчин). Каждая хромосома в паре является гомологичной другой хромосоме в паре.
В норме человек имеет диплоидный набор хромосом, то есть в каждой клетке содержится двойной комплект каждой из 23 хромосом.
Но есть ситуации, в которых клетки содержат ненормальный, не кратный 46, набор хромосом, что называется анеуплоидией. Анеуплоидия может выражаться, например, в наличии добавочной хромосомы (n + 1, 2n + 1 и т. п.) или в нехватке какой-либо хромосомы (n — 1, 2n — 1 и т. п.).
Формы анеуплоидии:
- моносомия (наличие одной из пары хромосом, например, синдром Шерешевского-Тернера, выражающийся в наличие одной половой Х-хромосомы)
- трисомия (наличие трех вместо 2 хромосом пары).
- тетрасомия (4 гомологичные хромосомы вместо пары в диплоидном наборе)
- пентасомия (5 вместо 2-х) встречаются чрезвычайно редко.
Дальше речь пойдет о самых частых хромосомных аномалиях — трисомиях. В некоторых случаях дополнительная хромосома представлена целой отдельной хромосомой (полная трисомия), а в некоторых этот генетический материал переносится на другую хромосому, что называют транслокацией.
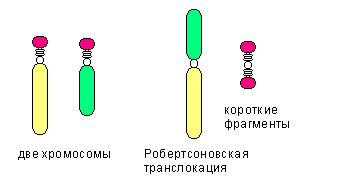
Среди транслокаций также выделяют:
- реципроктную транслокацию, когда неготомологичные хромосомы обмениваются участками
- робертсоновскую транслокацию (см.рис), при которой две неготомологичные хромосомы объединяются в одну.
- Сбалансированная транслокация не сопровождается утратой генетического материала.
Мозаицизмом называют ситуацию, когда среди всех клеток организма есть нормальные, а есть клетки с патологией (например, с трисомией). В этом случае степень отклонений зависит от количества клеток, которые имеет ненормальный генетический материал.
Хромосомы в случае синдрома Патау – Трисомия 13
Хромосомы в случае синдрома Эдвардса – Трисомия 18
Факторы риска
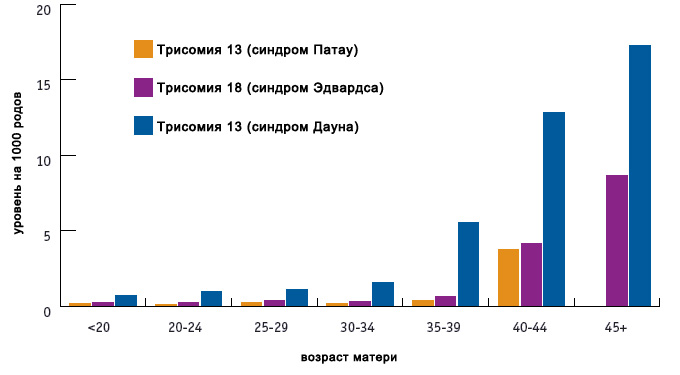
Основными факторами риска являются возраст (особо значимо для синдрома Дауна), а также воздействие радиации, некоторых тяжелых металлов. Следует учитывать, что даже без факторов риска плод может иметь патологию.
Как видно из графика, зависимость величины риска от возраста наиболее значима для синдрома Дауна, и менее значима для двух других трисомий.
Синдром Эдвардса
Синдром Эдвардса характеризуется трисомией по 18 хромосоме и комплексом множественных пороков развития.
В одном случае из 10 наблюдается мозаицизм, то есть лишняя хромосома есть не во всех клетках организма. Возможна и частичная трисомия с присоединением части 18 хромосомы к другой хромосоме.
Во время беременности наблюдается малый вес плода, многоводие, небольшая плацента и наличие одной артерии плаценты.
Новорожденные имеют изменение формы черепа, маленькие рот и целюсть, лицевой дисфорфизм, дефекты глаз и низкие деформированные ушные раковины. Также наблюдаются численные аномалии пальцев рук и ног, деформация стопы («стопа-качалка»).
Из дефектов внутренних органов наиболее часто встречаются пороки сердца и сосудов. У всех наблюдается гипоплазия мозжечка.
Синдром Эдвардса характеризуется умственной отсталостью и задержкой в развитии.
Большая часть детей умирает в первые месяцы жизни.
Синдром Патау
Синдром Патау обусловлен наличием лишней 13 хромосомы.
Это заболевание встречается примерно 1 на 5000-10000 родов. Частота встречаемости меняется в связи с возможностями пренатального скрининга и диагностики. Большая часть детей умирают в первые недели/месяцы жизни.
Дети с синдромом Патау небольшого роста, с микроцефалией, имеют покатый лоб, суженные глазные щели, микрофтальмия, миеломенингоцеле, помутнение роговицы, запавшая переносица и широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, короткая шея, флексорное положение кистей, сморщенная кожа задней поверхности шеи. Характерна умственная отсталость. Внутренние органы имеют дефекты: пороки сердца, сосудов, поджелудочной железы, селезенки, почек.
Во время беременности в большинстве случаев наблюдается многоводие.
Синдром Патау может быть обусловлен как простой трисомией, так и робертсоновской транслокацией. Мозаицизм и неробертсоновская транслокация встречаются редко.
Источник