
Дети с синдромом каудальной регрессии

Синдром каудальной регрессии – редкий тяжелый врожденный порок развития дистального отдела позвоночника и спинного мозга. В иностранной литературе встречается несколько терминов, обозначающих данное патологическое состояние: сакральная или люмбосакральная агенезия, синдром каудальной дисплазии, каудальная дисгенезия. Клиническая картина (см. далее) заболевания сопровождается гипоплазией нижней половины туловища и конечностей вследствие грубого порока развития каудального отдела позвоночника и спинного мозга. В зависимости от уровня и тяжести поражения последнего наблюдается различная степень выраженности неврологического дефицита. В зависимости от уровня поражения позвоночника могут отсутствовать копчиковые, крестцовые, поясничные и даже нижнегрудные позвонки, что определяет вариант порока.
Крайне тяжелая форма каудальной регрессии называется сиреномелией, или синдромом русалки. Частота встречаемости этого летального порока – 1 на 60 тыс. новорожденных. Патогномоничным признаком данной аномалии, впервые описанной в 1961 г. Duhamel , является слияние нижних конечностей. Сращение может быть костным или в пределах мягких тканей. В большинстве слчаев сиреномелии наблюдаются агенезия почек, слепо оканчивающаяся толстая кишка, отсутствие наружных и внутренних гениталий, единственная пупочная артерия, атрезия ануса.
Этиология синдрома каудальной регрессии окончательно не выяснена. Большинство авторов причинными факторами в генезе данной патологии рассматривают сахарный диабет у матерей, генетическую предрасположенность и недостаточное кровоснабжение нижней половины тела плода – установлено, что у плодов с данной патологией кровь шунтируется через аномальный сосуд в плаценту, не осуществляя кровоснабжение каудальных структур.
Классификация синдрома каудальной регрессии. С учетом накопленного опыта и различных клинических форм первая классификация синдрома каудальной регрессии предложена в 1978 г. Renshaw, который описал четыре варианта порока:
- Тип 1 — полная или частичная односторонняя сакральная агенезия: тазовое кольцо и люмбосакральное сочленение интактны, отсутствует позвоночно-тазовая нестабильность. Односторонняя агенезия крестца приводит к перекосу таза и поясничному сколиозу, который обычно не прогрессирует и не требует хирургического лечения. У больных с типом 1 может встречаться эквиноварусная деформация стоп и неврологический дефицит в виде выпадения чувствительности в соответствии с поражением крестцовых корешков.
- Тип 2 — неполная сакральная агенезия с частичным, но двухсторонним дефектом, стабильным суставом между подвздошными костями и нормальным или гипопластичным S1 позвонком. Позвоночно-тазовое сочленение у пациентов данной группы обычно стабильно, но часто встречаются врожденные аномалии развития позвонков на фоне нарушения их формирования и слияния (полупозвонки, клиновидные и бабочковидные позвонки). Деформация позвоночника у таких пациентов приобретает прогрессирующий характер и требует хирургической коррекции. В неврологической картине отмечаются парезы или параличи нижних конечностей без нарушения чувствительности. Деформации коленных суставов и стоп выражены умеренно. Большинство больных передвигается самостоятельно.
- Тип 3 — вариабельная поясничная и полная крестцовая агенезия, при которой подвздошные кости соединяются с боковыми поверхностями последнего позвонка. У пациентов данной группы позвоночно-тазовое сочленение относительно стабильное, несмотря на отсутствие в некоторых случаях L5 позвонка. В процессе роста и развития у ребенка наблюдаются прогрессирующий кифоз или сколиоз. Неврологический статус — двигательные нарушения с уровня вертебрального дефекта. При полном отсутствии крестца ягодицы плоские, а межъягодичная складка укорочена. Отмечаются вывихи бедер, контрактуры коленных суставов и деформация стоп, требующие оперативного лечения. Пациенты не могут передвигаться без ортезов и костылей.
- Тип 4 — вариабельная поясничная и полная сакральная агенезия, когда каудальная пластинка самого нижнего позвонка располагается над сочленением подвздошных костей. Этот тип представляет собой классическую форму синдрома каудальной регрессии или люмбосакральной агенезии. Для пациентов характерно положение в позе Будды, низкий рост, выраженная диспропорциональность грудной клетки и таза. Имеется отчетливая нестабильность таза. В положении пациента сидя таз максимально приближен к грудной клетке спереди. Почти у всех пациентов развивается кифотическая или кифосколиотическая деформация. Движения в тазобедренных суставах резко ограничены из-за выраженных сгибательно-отводящих контрактур. Сгибательные контрактуры коленных суставов часто сочетаются с характерными парусовидными подколенными складками, так называемыми птеригиумами. Как правило, наблюдаются фиксированные эквиноварусные деформации стоп.
В 1996 г. Cama et al. (1996) детально изучили группу пациентов с синдромом каудальной регрессии и выделили пять категорий:
- полная агенезия крестца с нормальным или уменьшенным поперечным диаметром таза и возможным отсутствием нескольких поясничных позвонков;
- полная сакральная агенезия;
- частичная сакральная агенезия или гипоплазия крестца (сохранен S1 позвонок);
- полукрестец (hemisacrum);
- копчиковая агенезия.
В 2002 г. Guelle et al. (2002) предложили новую классификацию синдрома каудальной регрессии, связывающую клинико-рентгенологический тип порока с потенциальной возможностью самостоятельной ходьбы. На основании опыта лечения 18 пациентов выделены две группы: у пациентов первой группы (13 наблюдений) присутствовала только люмбосакральная агенезия; второй группы (5 наблюдений) — люмбосакральная агенезия сочеталась с миеломенингоцеле. В каждой из групп отмечено три типа деформации позвоночника:
- тип А — небольшой дефект меж у подвздошными костями или сращение подвздошных костей по средней линии; отсутствие одного или нескольких поясничных позвонков; каудальный позвонок сочленяется с тазом по средней линии;
- тип B — полное сращение подвздошных костей, отсутствие нескольких поясничных позвонков, каудальный позвонок сочленяется с одной из подвздошных костей;
- тип С — полное сращение подвздошных костей между собой, отсутствие всех поясничных позвонков, значительный дефект между интактным грудным позвонком и тазом.
Все пациенты первой группы с деформациями типа А и частично типа В имели хорошую перспективу к самостоятельной ходьбе после хирургической коррекции деформаций нижних конечностей. Оставшиеся больные (вторая группа; тип С и частично тип В первой группы) самостоятельно передвигаться не могли, показаниями к корригирующим вмешательствам являлись невозможность сидения и/или ношения ортопедической обуви и ортезов.
Клиника. У пациентов с синдромом каудальной регрессии отмечается низкий рост, что обусловлено укорочением туловища и конечностей. Наиболее яркими клиническими проявлениями данного синдрома являются сужение и гипоплазия таза, гипотрофия нижних конечностей, врожденные вывихи бедер, сгибательные контрактуры тазобедренных и коленных суставов, эквиноварусные деформации стоп. В большинстве описанных наблюдений данная аномалия сочетается с пороками других органов и систем, что требует привлечения к лечению пациентов специалистов различного профиля. Данный врожденный дефект может сопровождаться рядом аномалий со стороны:
- центральной нервной системы (миеломенингоцеле, гидроцефалия, мальформация Арнольда-Киари, голопрозэнцефалия),
- сердца (дефект межжелудочковой перегородки);
- желудочно-кишечного тракта – трахео-эзо-фагеальный свищ, дефект передней брюшной стенки, паховая грыжа, мальротация кишечника, атрезия двенадцатиперстной кишки, атрезия прямой кишки;
- мочеполового тракта – уретеро-гидронефроз, мочепузырный рефлюкс, экстрофия мочевого пузыря, ректовагинальный и ректоуретеральный свищи, подковообразная почка, гипоспадия, атрезия уретры, транспозиция наружных гениталий, крипторхизм).
В неврологической картине заболевания наблюдаются глубокиепарезы или плегия нижних конечностей, мышечная гипотония, угнетение сухожильных рефлексов. Выраженность неврологического дефицита прямо коррелирует с уровнем обрыва спинного мозга и корешков. Нередко агенезия какого‑либо отдела позвоночника входит в состав генетических синдромов: OEIS-комплекс (омфалоцеле, экстрофия клоаки, атрезия ануса, пороки развития крестца), VATER‑синдром (вертебральные аномалии, атрезия ануса, трахеопищеводный свищ, атрезия пищевода, аномалии почек).
Лечение. Подход к лечению пациентов с синдромом каудальной регрессии должен быть комплексным, этапным и выбор в пользу хирургического или консервативного лечения решается строго индивидуально. Ведение данной группы пациентов ставит перед врачом ряд кардинальных задач: устранение позвоночно-тазовой нестабильности, коррекцию деформаций нижних конечностей, лечение осложнений, связанных с пороками развития других органов и систем с привлечением соответствующих специалистов.Тактика ведения пациентов с позвоночно-тазовой нестабильностью различна. Многие авторы придерживаются активной хирургической тактики и считают наличие нестабильности показанием к реконструктивной операции и инструментальной фиксации с целью освобождения верхних конечностей как опоры для нестабильного туловища, защиты внутренних органов от компрессии и деформации, создания стабильного позвоночно-тазового комплекса. В целом, отмечены положительные результаты позвоночно-тазового соединения.
В оперативном лечении вывихов бедер применяется раннее открытое вправление бедра, подвертельная корригирующая остеотомия бедренной кости с остеотомией таза или без нее. Сгибательные контрактуры коленных суставов представляют особые трудности для хирургического лечения и склонны к рецидивам даже после полной коррекции. Для устранения контрактур коленных суставов используют задний релиз с Z-образной пластикой кожной подколенной складки с наложением дистракционного аппарата и постепенным разгибанием сустава в сочетании с надмыщелковой разгибательной остеотомией бедренной кости. Guille et al. считают, что корригирующие операции на нижних конечностях должны выполняться у пациентов, потенциально способных к самостоятельному передвижению. У остальных эти операции применяют с целью облегчения сидения в коляске, ношения обуви и ортезов. Для устранения деформаций стоп и придания им опороспособности выполняют трехсуставной артродез с последующим снабжением ортезами. В отношении показаний к ампутациям нижних конечностей многие авторы считают возможным выполнение двусторонней подвертельной ампутации или дезартикуляции на уровне коленных суставов при тяжелых деформациях с последующим протезированием нижних конечностей, что позволяет пациенту уверенно сидеть без опоры на верхние конечности и самостоятельно передвигаться.
Литература: 1. статья «Cиндромом каудальной регрессии» С.В. Виссарионов, И.В. Казарян (Научно-исследовательский детский ортопедический институт им. Г.И. Турнера, Санкт-Петербург), статья опубликована в журнале «ХИРУРГИЯ ПОЗВОНОЧНИКА» 2/2010 (С. 50–55) [►]. 2. статья «Лечение пациентов с синдромом каудальной регрессии» С.В. Виссарионов, И.В. Казарян, С.М. Белянчиков (Научно-исследовательский детский ортопедический институт им. Г.И. Турнера, Санкт-Петербург), статья опубликована в журнале «ХИРУРГИЯ ПОЗВОНОЧНИКА» 3/2011 (С. 56–59) [►].
Источник
Синдром каудальной регрессии (сакральная или люмбосакральная агенезия, синдром каудальной дисплазии, каудальная дисгенезия, сиреномелия) — редкий врожденный порок развития дистального отдела позвоночника и спинного мозга. Впервые врожденный дефект в виде агенезии дистальной части позвоночника описан в 1852 году.
28842 •
•
21.02.2019
Иллюстрация Георгия Сапего
В целом, синдром каудальной регрессии – широкое понятие, которое относится к гетерогенной группе врожденных каудальных аномалий, затрагивающих хвостовой отдел позвоночника и спинной мозг, заднюю кишку, мочеполовую систему и нижние конечности.
До настоящего времени точные причины возникновения синдрома каудальной регрессии неизвестны. Как правило, большинство случаев являются спорадическими. Предполагается, что это мультифакториальный порок, т.е. в его возникновении могут принимать участие много факторов: как генетическая предрасположенность, так и средовые факторы. Возможно, что порок может возникать в результате нарушения кровообращения нижней половины тела плода. Одним из наиболее достоверных факторов риска является сахарный диабет матери. Примерно от 15 до 25% матерей детей с каудальной дисгенезией страдают инсулинозависимым сахарным диабетом. Есть указания на участие в развитии каудальной регрессии гена VANGL1. Этот ген имеет большое значение в раннем развитии эмбриона.
Частота каудальной регрессии оценивается как 1 – 5 случаев на 100000 рождений.
Клинические проявления синдрома отличаются выраженной вариабельностью. В результате выделяются различные типы нарушений каудального отдела. В некоторых случаях у пациентов может быть только частичная агенезия крестцового отдела. В других случаях крестец может отсутствовать полностью. Агенезия крестцового отдела сопровождается недоразвитием бедер, гипоплазией ягодичных мышц, наличием ямки в нижней части спины (крестцовая ямка или крестцовый синус). В более тяжелых случаях в процесс вовлекаются и поясничные позвонки. Чем выше поражение, тем тяжелее клинические проявления.
Аномальное развитие каудальной области позвоночника может вызвать дополнительные нарушения, затрагивающие спинной мозг и нижние конечности. В некоторых случаях может произойти нарушение или повреждение нижней части спинного мозга, вызывающее различные неврологические нарушения, включая нарушение функции мочевого пузыря и кишечника, увеличение частоты мочеиспускания и невозможность полного опустошения мочевого пузыря (нейрогенный мочевой пузырь).
Повреждение нервов также может привести к аномалиям нижних конечностей. Такие нарушения могут включать сгибательные контрактуры колена и бедра. Контрактура – это состояние, при котором сустав становится постоянно неподвижным в согнутом или выпрямленном положении, полностью или частично ограничивая движение пораженного сустава.
Возможен широкий спектр дополнительных симптомов, включая аномалии почек, аномалии верхних позвонков, аномалии лица (расщелины губы и/или неба) и атрезии аноректального отдела.
В зависимости от степени тяжести поражения выделяют несколько типов заболевания. В 2002 г. была предложена новая классификация, связывавшая клинико-рентгенологический тип порока с потенциальной возможностью самостоятельной ходьбы.
тип А — небольшой дефект между подвздошными костями или сращение подвздошных костей по средней линии; отсутствие одного или нескольких поясничных позвонков;
тип B — полное сращение подвздошных костей, отсутствие нескольких поясничных позвонков;
тип С — полное сращение подвздошных костей между собой, отсутствие всех поясничных позвонков, значительный дефект между интактным грудным позвонком и тазом.
Самая тяжелая форма каудальной регрессии называется сиреномелией, или синдромом русалки. Однако некоторые авторы отмечают, что в литературе нет единого мнения относительно связи между синдромом каудальной регрессии и сиреномелией. В частности, есть мнение, что сиреномелия и синдром каудальной регрессии являются частью патогенетического спектра, обусловленного первичным дефицитом каудальной эмбриональной мезодермы.
Диагноз синдрома каудальной регрессии ставится, как правило, еще пренатально по результатам УЗИ, в остальных случаях он ставится почти сразу после рождения. После того, как выполнено УЗИ и диагноз подтвержден, новорожденных направляют на магнитно-резонансную томографию (МРТ) для определения тяжести случая.
Лечение симптоматическое и зависит от того, насколько серьезны проявления. В некоторых случаях ребенку могут понадобиться только специальная обувь, опоры для ног или костыли. Кроме того, рекомендуется лечебная физкультура. В более тяжелых случаях часто требуется хирургическое лечение. Как правило, лечение в таких случаях требует координированных усилий различных специалистов: педиатров, нейрохирургов, урологов, нефрологов, ортопедов. Прогноз зависит от тяжести заболевания.
Источник
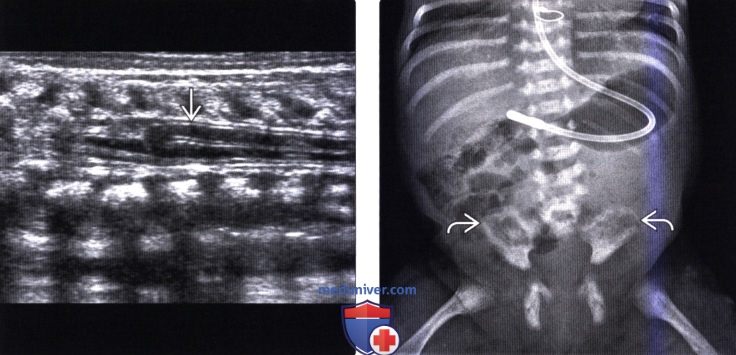
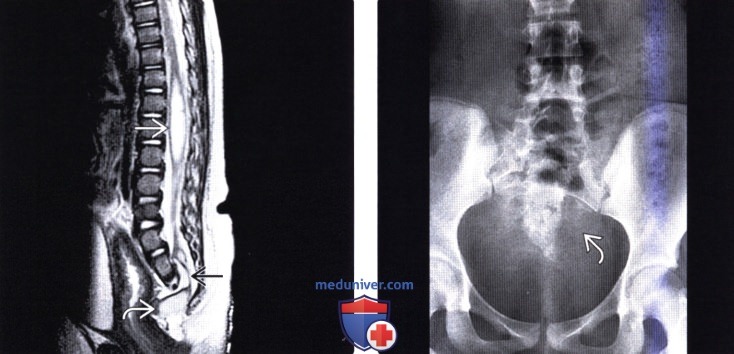
Лучевая диагностика синдрома каудальной регрессииа) Терминология: б) Визуализация: 1. Общие характеристики синдрома каудальной регрессии: 2. Рентгенологические данные: 3. КТ признаки синдрома каудальной регрессии: 4. МРТ признаки синдрома каудальной регрессии: 5. Ультразвуковые данные: 6. Несосудистые рентгенологические исследования: 7. Рекомендации по визуализации:
в) Дифференциальная диагностика синдрома каудальной регрессии: 1. Фиксированный спинной мозг: 2. Закрытая дизрафия позвоночника: 3. Скрытое крестцовое менингоцеле (СКМ): г) Патология: 1. Общие характеристики синдрома каудальной регрессии: 2. Стадирование, степени и классификация: 3. Макроскопические и хирургические особенности: д) Клинические особенности: 1. Клиническая картина синдрома каудальной регрессии: 2. Демография: 3. Течение заболевания и прогноз: 4. Лечение: е) Диагностическая памятка: ж) Список использованной литературы: – Также рекомендуем “МРТ при терминальном миелоцистоцеле (сирингоцеле)” Редактор: Искандер Милевски. Дата публикации: 18.7.2019 |
Источник