
Дети синдром секкеля фото детей

Что такое синдром Секкеля?
Синдром Секкеля чрезвычайно редкое наследственное заболевание, характеризующееся задержкой роста до рождения (задержка внутриутробного развития), что приводит к низкой массе тела при рождении. Задержка роста продолжается после рождения (послеродовой период), что приводит к низкорослости (карликовость). Другие симптомы и физические особенности, связанные с синдромом Секкеля, включают аномально маленькую голову (микроцефалия); разную степень умственной отсталости; и/или необычные характерные черты лица, включая «клювоподобный» выступ носа. Другие черты лица могут включать аномально большие глаза, узкое лицо, деформированные уши и/или необычно маленькую челюсть (микрогнатия). Кроме того, у некоторых младенцев может наблюдаться постоянная фиксация пятых пальцев в согнутом положении (клинодактилия), деформация (дисплазия) тазобедренного сустава, вывих кости в предплечье (радиальный смещение) и/или другие физические отклонения.
Признаки и симптомы
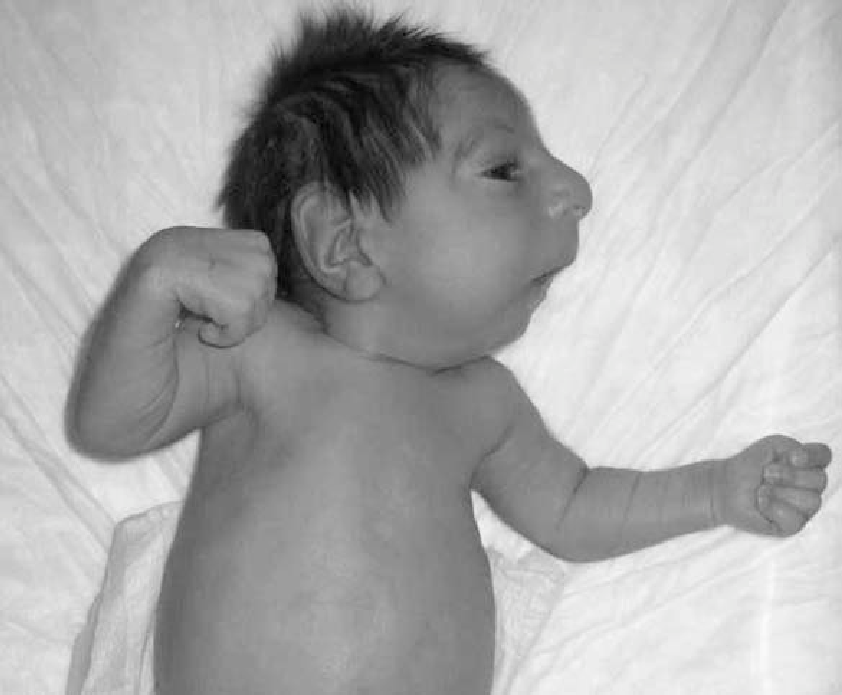
Синдром Секкеля характеризуется аномально медленным развитием плода и низкой массой тела при рождении. После рождения у ребенка будет наблюдаться медленный рост и созревание костей, что приведет к низкому, но пропорциональному росту (в отличие от карликовости коротких конечностей или ахондроплазии). Больные с синдромом Секкеля имеют различные физические отклонения и особенности развития, в том числе:
- Очень маленький размер и вес при рождении (в среднем 3,3 фунта или 1,4 кг);
- Чрезвычайно маленький, пропорциональный рост;
- Аномально маленький размер головы (микроцефалия);
- Клювовидный выступ носа;
- Узкое лицо;
- Деформированные уши;
- Необычно маленькая челюсть (микрогнатия);
- Умственная отсталость, часто тяжелая, с IQ (коэффициент интеллекта) менее 50.
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, пороки развития зубов и другие деформации костей. Также часто наблюдаются такие заболевания крови, как анемия (низкий уровень эритроцитов), панцитопения (недостаток клеток крови) или острый миелоидный лейкоз (тип рака крови).
В некоторых случаях у мужчин яички не могут опускаться в мошонку (крипторхизм), а у женщин может быть ненормально увеличенный клитор. Кроме того, у больных с синдромом может быть чрезмерное оволосение на теле и одна единственная глубокая складка на ладонях (известная как обезьянья складка).
Причины
Синдром Секкеля — чрезвычайно редкое заболевание, передающееся по аутосомно-рецессивному типу. Три варианта синдрома Секкеля включают нарушения или изменения (мутации) генов на трех разных хромосомах. Расположение генной карты: синдром Секеля 1 на хромосоме 3 (3q22-q24); Синдром Секкеля 2 на хромосоме 18 (18p11.31-q11) и синдром Секеля 3 на хромосоме 14 (14q21-q22).
Хромосомы, присутствующие в ядре клеток человека, несут генетическую информацию для каждого человека. Клетки человеческого тела обычно имеют 46 хромосом. Пары человеческих хромосом пронумерованы от 1 до 22, а половые хромосомы обозначены X и Y. У мужчин одна X и одна Y хромосома, а у женщин две X хромосомы. У каждой хромосомы есть короткое плечо, обозначенное буквой «p», и длинное плечо, обозначенное буквой «q». Хромосомы далее подразделяются на множество пронумерованных полос. Например, «хромосома 3q22-q24» относится к области между полосой 22 и полосой 24 на длинном плече хромосомы 3. Подобным образом «хромосома 18p11.31-q11.2» относится к более широкой области между полосой 11p31 на коротком плече хромосомы 18 и полосе 11.2 на длинном плече хромосомы 18. Опять же, хромосома 14q21-q22 относится к более узкой области на длинном отрезке хромосомы 14 между полосами 21 и 22. Пронумерованные полосы указывают расположение тысяч генов, присутствующих на каждой хромосоме.
Специфический ген, участвующий в синдроме Секкеля 1, известен как ген атаксии-телеангиэктазии и ген Rad3-родственного белка (ATR). Гены, участвующие в синдроме Секкеля 2 и 3 типов, неизвестны.
Генетические заболевания определяются комбинацией генов определенного признака, которые находятся на хромосомах, полученных от отца и матери.
Рецессивные генетические расстройства возникают, когда человек наследует один и тот же аномальный ген одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, он будет носителем болезни, но обычно бессимптомным. Риск для двух родителей-носителей передать дефектный ген и, следовательно, иметь больного ребенка, составляет 25% при каждой беременности. Риск иметь ребенка, который будет носителем, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей и будет генетически нормальным по данному признаку, составляет 25%. Риск одинаков для мужчин и женщин.
Все люди несут несколько аномальных генов. Родители, которые являются близкими родственниками (кровными родственниками), имеют больше шансов, чем неродственные родители, иметь один и тот же аномальный ген, что увеличивает риск рождения детей с рецессивным генетическим заболеванием.
Затронутые группы населения
Синдром Секкеля — чрезвычайно редкое наследственное заболевание, которое поражает в равной степени мужчин и женщин. Точная частота расстройства неизвестна. С момента первоначального описания в 1960 г. в медицинской литературе было зарегистрировано более 100 случаев.
Близкие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Секкеля. Сравнения могут быть полезны для дифференциальной диагностики:
Синдром Секкеля — один из шести расстройств в классе карликовости, известном как «изначальная карликовость». Эти расстройства имеют схожие характеристики, включая пороки развития скелета (дисплазию), дефицит роста до рождения (задержка внутриутробного развития) и в младенчестве и детстве, что в конечном итоге приводит к разной степени низкого роста. В эту группу заболеваний в настоящее время входят пять расстройств: синдром Секкеля, синдром Мейера-Горлина; Синдром Рассела-Сильвера; Остеодиспластическая примордиальная карликовость Маевского I/III; и остеодиспластическая примордиальная карликовость Маевского II типа.
Остеодиспластическая примордиальная карликовость Маевского II типа является чрезвычайно редким генетическим заболеванием, характеризующимся низким ростом, низкой массой тела при рождении, аномально маленькой головой (микроцефалия) и/или аномалиями скелета. Другие физические признаки могут включать большие глаза, выступающий нос в форме клюва, опущенную челюсть и/или узкое лицо. Обычно возникает серьезная недостаточность роста до рождения (внутриутробная недостаточность роста). В некоторых случаях может присутствовать умственная отсталость. Также могут появиться различные дополнительные симптомы. Конкретные симптомы и степень тяжести варьируются от случая к случаю. Считается, что II тип остеодиспластической примордиальной карликовости Маевского наследуется по аутосомно-рецессивному типу.
Диагностика
С появлением технически совершенной ультасонографии синдром Секкеля можно диагностировать до рождения (пренатально). При ультразвуковом исследовании плода используются отраженные звуковых волны для создания изображения развивающегося плода. После рождения можно заподозрить синдром Секкеля на основании тщательного клинического обследования, подробного анамнеза пациента и различных специализированных тестов. Хотя характерные черепно-лицевые, скелетные и/или другие физические аномалии, связанные с синдромом Секкеля, могут присутствовать при рождении, в некоторых случаях диагноз может не подтверждаться до тех пор, пока больной ребенок не вырастет и не разовьется полный синдром (например, когда станет очевидным низкий рост и/или умственная отсталость).
Низкий рост, связанный с синдромом Секкеля, предполагает пропорциональный рост рук и ног, что позволяет проводить дифференциальную диагностику от синдромов, связанных с низким ростом и аномально маленькими руками и ногами (карликовость с короткими конечностями).
Стандартные методы лечения
От синдрома Секкеля нет лекарства. Некоторые лекарства могут быть назначены для устранения других симптомов, связанных с заболеванием.
Лечение направлено на решение любой медицинской проблемы, которая может возникнуть, особенно заболеваний крови и структурных деформаций. Людям с умственными недостатками и их семьям потребуется соответствующая социальная поддержка и консультационные услуги.
Прогноз
Дети, страдающие синдромом Секкеля, могут жить в течение длительного периода времени, хотя они часто сталкиваются с глубокими психическими и физическими отклонениями.
Источник
Синдром Секкеля является унаследованной формой врожденной карликовости , а это значит, что младенец начинает очень мало и не может нормально расти после рождения. В то время как люди с синдромом Секкеля обычно будут пропорциональны по масштабу, у них будет отчетливо малый размер головы. Также распространена умственная отсталость.

Несмотря на множество физических и умственных проблем, с которыми сталкивается человек с синдромом Секкеля, многие из них, как известно, хорошо живут более 50 лет.
Причины синдрома Секкеля
Синдром Секкеля является наследственным расстройством, связанным с генетическими мутациями на одной из трех разных хромосом. Считается, что это бывает крайне редко, с немногим более 100 случаев, зарегистрированных с 1960 года. Многие дети, получившие диагноз синдрома Секкеля, родились у родителей, которые тесно связаны (родственники), например, брак с кузенами или братьями и сестрами.
Синдром Секкеля является рецессивным генетическим заболеванием, он возникает только тогда, когда ребенок наследует один и тот же ненормальный ген от каждого родителя. Если ребенок получает один нормальный ген и один ненормальный ген, ребенок будет носителем синдрома, но обычно не проявляет симптомов.
Если оба родителя имеют одинаковую хромосомную мутацию для синдрома Секкеля, их риск иметь ребенка с синдромом Секкеля составляет 25%, а риск наличия носителя — 50%.
Характеристики синдрома Секкеля
Синдром Секкеля характеризуется аномально медленным развитием плода и низким весом при рождении.
После рождения у ребенка будет медленный рост и созревание костей, что приводит к короткому, но пропорциональному росту (в отличие от ахондроплазии). Лица с синдромом Секкеля обладают различными физическими и характеристиками развития, в том числе:
Очень маленький размер и вес при рождении
- Чрезвычайно малый, пропорциональный рост
- Аномально небольшой размер головы (микроцефалия)
- Клювообразный выступ носа
- Узкое лицо
- Нарушенные уши
- Необычно маленькая челюсть (микрогнатия)
- Умственная отсталость, часто тяжелая с IQ менее 50
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, повреждение зубов и другие деформации кости. Расстройства крови, такие как анемия (низкие эритроциты), панцитопения (недостаточно клеток крови) или острый миелоидный лейкоз.
В некоторых случаях семенники у мужчин не спускаются в мошонку, а женщины могут иметь аномально увеличенный клитор. Кроме того, у людей с синдромом Секкеля могут быть чрезмерные волосы на теле и единая глубокая складка на ладонях (известная как складки обезьян).
Диагностика синдрома Секкеля
Диагноз синдрома Секкеля основан почти исключительно на физических симптомах. Возможно, потребуется рентгеновское изображение и другие инструменты (МРТ, КТ), чтобы отличить его от других подобных условий. В настоящее время нет лабораторных или генетических тестов, специфичных для синдрома Секкеля. В некоторых случаях окончательный диагноз не может быть сделан до тех пор, пока ребенок не станет старше и не появятся характерные симптомы.
Лечение синдрома Секкеля
Лечение синдрома Секкеля сосредоточено на любой медицинской проблеме, которая может возникнуть, особенно при нарушениях крови и структурных деформациях. У людей с психическими расстройствами и их семьям должна быть предоставлена соответствующая социальная поддержка и консультационные услуги.
Источник
Рождение детей с генетическими отклонения во все времена было тяжелым испытанием для их родителей. Если сегодня возможности врачей позволяют определять аномалии еще в период внутриутробного развития, то в XIX веке и практически на протяжении всего XX такие исследования были невозможными. Когда-то детей с визуальными или умственными дефектами стыдились. Их прятали или попросту избавлялись от них. Выжившим ничего не оставалось, как становиться актерами в сайдшоу и цирках фриков.
Последние как явление возникли в XVIII веке и обычно были частью развлекательной программы на ярмарках. В те времена людям не были интересны силачи или акробаты, владельцы такого «бизнеса» терпели нешуточные убытки. Публике необходимы были другие, более эмоциональные зрелища. Кому пришла в голову идея выставлять напоказ человеческое несовершенство, сейчас судить сложно. Но это оказалось востребованным.
В 1880-м году в Джорджии родилась необычная девочка, Минни Вусси. О ее детстве и молодости известно крайне мало, считалось, что в свое время она была «освобождена» из учреждения, в котором содержались психически больные люди. Она страдала от редкого врожденного дефекта, синдрома Вирхова-Секкеля, что сильно сказалось на ее внешности и навыках.
Минни была очень невысокой, с неправильным строением скелета.
Девочка несколько отставала в умственном развитии и при этом сильно отличалась от большинства людей. Небольшая голова с практически полным отсутствием волос и зубов, суженное кницу и напоминающее птичье лицо делали ее далеко не красавицей. К тому же, она была либо полностью слепа, либо сильно близорука.
Однако именно это стало ее билетом в мир развлечений. В различных сайдшоу Минни появлялась под псевдонимом Девушка-птица Ку-Ку. Она наряжалась в «птичьи» костюмы, ходила по сцене, размахивая руками, и разговаривала на никому не известном языке, который в народе называли просто тарабарщиной.
Когда точно и отчего умерла Девушка-птица Ку-Ку, была ли у нее личная жизнь, достоверно не известно. Последние данные гласили о том, что еще в 1960-м она выступала со своим «коронным» номером.
Хотя синдром Вирхова-Секкеля считается очень редким, последовательница у Минни нашлась и в наши дни. Она родилась с несколько похожим диагнозом. Девушка страдала от синдрома Халлермана-Штрайфа, ее рост составлял всего 144 см. Для этого заболевания также характерны черепные аномалии, из-за которых лицо напоминает птичье, проблемы с зубами, волосами и некоторые другие пороки развития.
Тем не менее Сара, не любившая говорить о своем состоянии и в свое время выступавшая на Паралимпиаде, стала востребованной артисткой. Девушка выступала в цирках и физических театрах, превращая недостатки в предмет восхищения. Обладая плохим зрением и прекрасно чувствуя свое тело, она демонстрировала публике невероятные номера, которые и для совершенно здорового человека довольно опасны.
Сара – активный адвокат людей с ограниченными возможностями. Девушка считала, что они не менее достойны быть частью современного искусства, чем другие.
В конце концов, многие сферы, включая моду и красоту, становятся все более инклюзивными. Уважения заслуживает уже то, что такие люди умеют принимать свою уникальность и превращать ее в достоинства, не так ли? Что по этому поводу думаете вы?
Понравилась статья? Подпишитесь на канал, чтобы быть в курсе самых интересных материалов
Подписаться
Источник
Дата публикации: 17.12.2018
Синдром Ангельмана – сложное генетическое заболевание, которое поражает нервную систему у детей, характеризуется микроцефалией (небольшим размером головы), эпилептическими расстройствами и нетипично веселым нравом. Патология вызывает задержку умственного развития ребенка, значительные нарушения речи и координации движений. Это неврологическое заболевание еще именуют «синдромом счастливой марионетки» из-за того, что больные дети много улыбаются, смеются, взмахивают руками. Какие еще признаки могут указывать на синдром Ангельмана? Лечится ли эта патология? Обратимся к консультации экспертов.
Причины возникновения патологии
Синдром Ангельмана получил свое название от Гарри Ангельмана – британского педиатра, который первым, в 1965 году, распознал и дифференцировал типичные признаки заболевания, многие из которых схожи по симптомам с аутизмом или церебральным параличом.
Болезнь встречается редко, диагностируется у одного из 10000-20000 детей. Оба пола одинаково восприимчивы к патологии. Причина – мутация гена Ube3a в 15 хромосоме у матери. Реже синдром вызывается наследованием двух копий этого гена от отца, хромосомной перестройкой (транслокацией), нарушением развития яйцеклетки.

Симптомы болезни
На сегодняшний день симптомы, сопутствующие заболеванию, четко определены. Задержка развития является первым симптомом, который становится очевидным к тому времени, когда новорожденному исполняется от 6 месяцев до 1 года. Другие – судороги, нарушение речи, умственная отсталость – сохраняются в течение всей жизни. Сразу после рождения дети с синдромом Петрушки (еще один неофициальный термин болезни Ангельмана в России) кажутся совершенно нормальными. Тем не менее, в течение первого месяца после рождения у детей уже становятся заметны проблемы с питанием.
Возможно, самая яркая характеристика болезни – это состояние счастья и удовлетворенности – уникальная поведенческая особенность, при которой больной ребенок или взрослый будет часто смеяться, улыбаться, сохраняя при этом повышенное настроение и возбудимость продолжительное время. Часто ходят с поднятыми локтями и согнутыми запястьями. Смех и улыбка получаются, порой, неуместными. Несмотря на свое физическое и интеллектуальное ограничение, дети имеют безграничное любопытство и постоянно находятся в движении. Они наслаждаются игрой и общением с людьми, проявляют глубокое желание личного общения и привязанности, с удовольствием позируют на фото.

Итак, перечислим признаки синдрома Ангельмана:
- гиперактивность;
- малая окружность головы;
- широко расставленные глаза/косоглазие;
- проблемы со сном (не более 4-5 часов за один раз);
- тяжелые нарушения речи;
- гипотония (снижение мышечного тонуса);
- аномальное увеличение размера рта и дефицит роста верхней челюсти;
- судороги (проявляются при достижении детьми возраста 2-3 лет);
- специфическое поведение (постоянная улыбка на лице, смех без причины, взмахи руками);
- симптомы синдрома дефицита внимания;
- атаксия (нарушение баланса и координации движений);
- чувствительность к жаре;
- тяга к воде и блестящим предметам;
- грубые черты лица (выпуклая голова, широкие губы и язык, маленькие деформированные зубы);
- неестественно светлая кожа и волосы;
- сколиоз;
- плоский затылок;
- гладкие ладони со складками;
- эпилептические припадки.

Осложнения, связанные с синдромом Ангельмана
У большинства детей с этим расстройством наблюдаются:
- Повышенная возбудимость. Больному ребенку трудно сосредоточиться на конкретной деятельности в течение определенного периода времени. Неспособность сконцентрироваться заставляет их переходить от одного занятия к другому. Фактически они не могут справиться с простыми задачами, не могут играть с определенными игрушками. Засовывают пальцы себе в рот во время игры или занятий.
- Повышенный аппетит. У детей старшего возраста и подростков отмечается обжорство, которое приводит к избыточному весу.
- Искривление позвоночника. При достижении зрелого возраста у пациентов возникает деформация позвоночника с боковым искривлением.
- Трудности со сном. Дети с синдромом Ангельмана испытывают меньшую потребность во сне, чем обычные дети.
- Проблемы с питанием. У младенцев и детей постарше могут возникнуть проблемы с глотанием и сосанием, что мешает их росту и развитию.
По мере взросления некоторые из этих форм поведения смягчаются, а гиперактивность и плохой сон могут полностью исчезнуть.

Лечение
Синдром Ангельмана – неизлечимая болезнь, но сегодня существует множество вариантов поддерживающей терапии. Благодаря ей интенсивность некоторых симптомов значительно уменьшается. Справиться с патологией помогают:
- противосудорожные препараты, которые уменьшают количество эпилептических приступов и позволяют управлять ими;
- мелатонин, улучшающий и продлевающий сон;
- лекарства для профилактики гастроэзофагеального рефлюкса у детей раннего возраста;
- поведенческая и коммуникативная терапия, повышающие качество жизни больных;
- трудотерапия для обучения самообслуживания (для больных детей предпочтительна одежда без застежек, пуговиц, обувь без шнурков);
- физиотерапия;
- хирургическое вмешательство при косоглазии;
- ношение брекетов.
Синдром Ангельмана не является прогрессирующим заболеванием. При должной терапии симптомы уменьшаются, но в любом случае больные нуждаются в пожизненном уходе.
Если вы заметили у своего ребенка странности в развитии и поведении, обсудите это со своим педиатром. При осмотре ребенка специалист должен проявить осторожность, так как высока вероятность постановки ошибочного диагноза. Среди особенностей врач должен обратить внимание в первую очередь:
- на характерные аномалии головы и лица;
- счастливую улыбку;
- пропущенные или отсроченные этапы развития ребенка, особенно отсутствие речи;
- моторную дисфункцию (мелкую дрожь, взмах рук, жесткую походку);
- историю развития с судорогами.

Безусловно, ранняя диагностика позволит использовать все терапевтические возможности, которые помогут улучшить жизнь вашего ребенка!
Похожие посты
Оставить комментарий
Источник