
E74 код по мкб

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
E74,0 Болезни накопления гликогена.
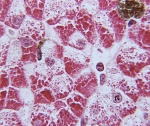
E74.0 Болезни накопления гликогена
Синонимы диагноза
Болезни накопления гликогена, гликогенозы, помпе болезнь.
Описание
Болезнь Помпе. Редкая наследственная патология, одна из форм лизосомных болезней накопления, характеризующаяся нарушением процессов расщепления гликогена в нервных и мышечных клетках (скелетные мышцы, миокард). Симптомы заболевания довольно вариабельны по времени своего проявления и выраженности у разных больных, традиционно наблюдается прогрессирующая мышечная слабость, при некоторых формах – кардиомегалия с дилятационной кардиомиопатией. Диагностика болезни Помпе производится на основании данных наследственного анамнеза, гистологического и гистохимического изучения мышечных тканей, биохимического анализа крови, а также генетических исследований. Лечение в настоящий момент может производиться с помощью фермент-заместительной терапии, однако эффективность этой методики неодинакова у разных пациентов.
E74.0 Болезни накопления гликогена
Дополнительные факты
Болезнь Помпе (гликогеноз 2-го типа, недостаточность кислой альфа-глюкозидазы) – наследственное заболевание, при котором из-за нарушения процессов обмена гликогена происходит повреждение нервных и мышечных тканей. Впервые было описано в 1932 году голландским ученым И. Помпе, с тех пор официально зарегистрировано более 50 случаев патологии. Болезнь Помпе с равной степенью вероятности поражает как мужчин, так и женщин, встречаемость колеблется от 1:60000 (взрослая форма) до 1:140000 (ранняя, или инфантильная форма). Является одной из немногих лизосомных болезней накопления, в отношении которой было разработано эффективное специфическое лечение, одобренное в США в 2006 году и в России – в 2013 г. Однако стоимость этиотропной терапии болезни Помпе крайне высока и составляет несколько сотен тысяч долларов в год. Смертность в случае отсутствия лечения зависит от формы патологии – детская инфантильная форма часто приводит к летальному исходу на 1-2-м году жизни ребенка, при типе болезни с отсроченным началом нарастание симптомов идет намного медленнее.
Причины
Болезнь Помпе является классическим гликогенозом, при ней в тканях скелетных мышц, миокарда и отчасти нервной системы формируются отложения гликогена по причине невозможности его расщепления. Это происходит в результате мутации гена GAA, расположенного на 17-й хромосоме – он кодирует последовательность кислой альфа-1,4-глюкозидазы или мальтазы. Это один из ключевых ферментов лизосом, участвующий в расщеплении молекулы гликогена на более простые отрезки, которые, в конечном итоге, деградируют до глюкозы, вступающей в энергетический обмен клетки. Так как гликоген является важным депо энергии для таких структур, как скелетные мышцы, миокард, печень и нервная ткань, проявления болезни Помпе сводятся именно к патологическим изменениям данных органов.
В результате подобных изменений сначала возникает дефицит энергии в клетках – потребности тканей в глюкозе покрываются только за счет ее поступления из крови. Кроме того, в лизосомах при болезни Помпе начинает накапливаться гликоген, формируя крупные включения в виде вакуолей, в дальнейшем приводя к дистрофии и повреждению клеток. Наследование дефектных вариантов гена GAA происходит по аутосомно-рецессивному типу. Наличие нескольких форм заболевания предположительно объясняется разными типами мутаций вышеуказанного гена. Возможно, при некоторых дефектах происходит не полное исчезновение, а лишь снижение активности кислой альфа-1,4-глюкозидазы, что и приводит к более позднему развитию болезни Помпе и медленному прогрессированию заболевания. Определение формы патологии играет важную роль для составления ее прогноза и схемы лечения.
Классификация
На сегодняшний день специалисты выделяют несколько основных форм болезни Помпе, основное различие между которыми заключается в сроках начала заболевания и выраженности симптомов. В большинстве случаев, с гликогенозом 2-го типа сталкиваются врачи-педиатры, однако имеется тип заболевания, выявляемый у взрослых.
• Ранняя инфантильная форма. Такая разновидность болезни Помпе считается наиболее тяжелой. Выявляется еще на первых месяцах жизни, симптомы миопатии, поражения печени (гепатомегалия) и сердца (кардиомиопатия) достаточно быстро прогрессируют. Обычно больные с ранней инфантильной формой болезни Помпе умирают от сердечной или дыхательной недостаточности в возрасте до года.
• Поздняя инфантильная форма. Первые симптомы возникают в возрасте 1 – 3 года, скорость их прогрессирования также намного медленнее. При данном типе болезни Помпе наиболее выражены поражения миокарда, смерть наступает к подростковому возрасту от сердечной недостаточности.
• Ювенильная форма болезни Помпе развивается в возрасте 6-10 лет. Так же, как и в предыдущем варианте, основным органом-мишенью болезни становится сердце, смерть от нарастающей сердечной недостаточности наступает к 20 годам.
• Взрослая форма болезни Помпе. Манифестация симптомов происходит в 20 – 40 лет. Ведущим симптомом является медленно прогрессирующая миопатия, поражения печени практически никогда не регистрируются, в некоторых случаях возможны незначительные нарушения миокарда. При этом типе болезни Помпе больные во многих случаях доживают до старости, лишь иногда возможен более ранний летальный исход из-за дыхательной или сердечной недостаточности.
Методами современной генетики на сегодняшний момент не определена взаимосвязь между отдельными типами мутаций гена GAA и формами болезни Помпе. Возможно, причина такой вариабельности проявлений лежит совсем в другом – в литературе описаны семейные случаи заболевания, когда у родственников регистрировались различные формы патологии. Изучение закономерностей, приводящих к развитию болезни Помпе определенного типа, сильно осложняется относительной редкостью данного синдрома.
Симптомы
Проявления болезни Помпе довольно сильно отличаются при различных формах заболевания. Ранний инфантильный тип характеризуется выраженной мышечной слабостью младенца, снижением его двигательной активности, плаксивостью. В педиатрии при осмотре такого больного часто выявляется задержка психомоторного развития, различная степень увеличения печени, пальпация иногда выявляет гипертрофию мышц, которые, однако, при этом довольно слабые. При дальнейшем развитии болезни Помпе возникают проблемы с кормлением из-за слабости сосательной мускулатуры, выявляется дисфагия и, как итог всего этого – гипотрофия. Нарастающая кардиомиопатия и слабость дыхательной мускулатуры со временем приводят к смерти ребенка.
Поздняя инфантильная и ювенильная формы болезни Помпе протекают практически одинаково, различается только срок появления симптомов патологии. Как правило, выявляется мышечная слабость, признаки кардиомиопатии. Со временем начинает формироваться выраженная дистрофия скелетной мускулатуры, кардиомегалия, на этом фоне начинают увеличиваться печень и селезенка. Длительность течения этих форм болезни Помпе составляет около 10-12 лет, после чего, при отсутствии лечения, наступает летальный исход из-за декомпенсированной сердечной недостаточности. Косвенным симптомом будет являться большая частота простудных заболеваний с легочными осложнениями, ночное апноэ, головные боли по утрам.
Диагностика
Выявление болезни Помпе можно производить многочисленными клиническими методиками – биохимическим анализом крови, изучением биоптата мышц, культур фибробластов или лейкоцитов больного, традиционными исследованиями (осмотр, ЭКГ, ЭхоКГ). При осмотре часто обнаруживается слабость и дистрофия мышц, при вовлечении в патологический процесс внутренних органов – гепато- и спленомегалия. На электрокардиограмме регистрируется укорочение интервала PQ, расширение комплекса QRS, обусловленного увеличением размеров миокарда. По этой же причине увеличивается длительность фазы реполяризации желудочков, что проявляется инверсией зубца T. Эхографическое исследование сердца показывает резкое увеличение его размеров за счет значительного утолщения стенок желудочков. При взрослой форме болезни Помпе вышеуказанные изменения миокарда могут не выявляться.
Биохимический анализ крови позволяет обнаружить как специфические, так и косвенные признаки заболевания. Специфическим исследованием будет определение активности кислой альфа-1,4-глюкозидазы в плазме крови, которая при болезни Помпе будет резко снижена. Косвенным указанием на наличие патологии является резкое повышение активности креатинфосфокиназы, обусловленное поражением мышечной ткани. Гистологическое исследование биоптата мышц выявляет в миоцитах многочисленные включения гликогена, часто придающие им вид «пенистых клеток». Гистохимическое изучение при болезни Помпе выявляет резкое снижение активности кислой альфа-1,4-глюкозидазы в мышечной ткани, фибробластах и лейкоцитах.
Врачом-генетиком может быть проведено генетическое определение болезни Помпе – оно производится методом прямого секвенирования последовательности гена GAA с целью выявления дефектных участков. Кроме того, может помочь в диагностике заболевания и составление наследственного анамнеза. Генетическая диагностика болезни Помпе включает в себя секвенирование GAA и у фенотипически здоровых родственников больного с целью выявления носительства патологического гена.
Лечение
На сегодняшний день единственным методом специфического лечения болезни Помпе является фермент-заместительная терапия с целью восполнения дефицита кислой альфа-1,4-глюкозидазы. Для этого используют препарат альфа алглюкозидазы производства США. Стоимость этого лечения крайне высока (годовой курс стоит 100-400 тысяч долларов), однако его эффективность неодинакова у разных больных. Других способов лечения болезни Помпе в настоящее время не существует.
Прогноз
Без лечения при инфантильных и ювенильной формах заболевания прогноз неблагоприятный, у взрослого типа – неопределенный. Профилактика возможна только путем своевременного выявления носительства болезни Помпе (в случае наличия патологии у кровных родственников) и последующей генетической пренатальной диагностики.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Профилактика
Названия
Название: E74,2 Нарушения обмена галактозы.
Описание
Галактоземия. Наследственная ферментопатия, характеризующаяся нарушением нормального процесса углеводного обмена, а именно – метаболизма галактозы. Признаками галактоземии являются непереносимость грудного молока и молочных смесей, рвота, анорексия, гипотрофия, желтуха, цирроз печени, спленомегалия, отеки, катаракта, задержка психомоторного развития. Скрининг на галактоземию проводится всем новорожденным; дополнительное обследование включает определение уровня галактозы в крови и моче, проведение нагрузочных проб с галактозой и глюкозой, генетическое тестирование, УЗИ брюшной полости, ЭЭГ и тд Основу терапии галактоземии составляет безлактозная диета, назначаемая с первых дней жизни ребенка.
Дополнительные факты
Галактоземия – наследственная патология обмена веществ, обусловленная недостаточностью активности ферментов, принимающих участие в метаболизме галактозы. Неспособность организма утилизировать галактозу приводит к тяжелым поражениям пищеварительной, зрительной и нервной системы детей в самом раннем возрасте. В педиатрии и генетике галактоземия относится к редким генетическим заболеваниям, встречающимся с частотой один случай на 10 000 – 50 000 новорожденных.
Впервые клиника галактоземии была описана в 1908 году у ребенка, страдавшего сильным истощением, гепато- и спленомегалией, галактозурией; при этом заболевание исчезло сразу после отмены молочного питания. Позднее, в 1956 г. Ученый Герман Келкер определил, что в основе заболевания лежит нарушение метаболизма галактозы.
Причины
Галактоземия является врожденной патологией, наследуемой по аутосомно-рецессивному типу, т. Е. Заболевание проявляется только в том случае, если ребенок наследует две копии дефектного гена от каждого из родителей. Лица, гетерозиготные по мутантному гену, являются носителями заболевания, однако у них тоже могут развиваться отдельные признаки галактоземии в легкой степени.
Превращение галактозы в глюкозу (метаболический путь Лелуара) происходит при участии 3-х ферментов: галактоза-1-фосфатуридилтрансферазы (GALT), галактокиназы (GALK) и уридиндифосфат-галактозо-4-эпимеразы (GALE). В соответствии с дефицитом этих ферментов различают 1 (классический вариант), 2 и 3 тип галактоземии.
Выделение трех типов галактоземии не совпадает с порядком действия ферментов в процессе метаболического пути Лелуара. Галактоза поступает в организм с пищей, а также образуется в кишечнике в процессе гидролиза дисахарида лактозы. Путь метаболизма галактозы начинается с ее превращения под действием фермента GALK в галактозо-1-фосфат. Затем при участии фермента GALT галактозо-1-фосфат преобразуется в УДФ-галактозу (уридилдифосфогалактозу). После этого с помощью GALE метаболит превращается в УДФ – глюкозу (уридилдифосфоглюкозу).
При недостаточности одного из названных ферментов (GALK, GALT или GALE) концентрация галактозы в крови значительно повышается, в организме накапливаются промежуточные метаболиты галактозы, которые вызывают токсическое поражение различных органов: ЦНС, печени, почек, селезенки, кишечника, глаз и тд Нарушение метаболизма галактозы и составляет суть галактоземии. Наиболее часто в клинической практике встречается классический (1 тип) галактоземии, обусловленный дефектом фермента GALT и нарушением его активности. Ген, кодирующий синтез галактоза-1-фосфатуридилтрансферазы, находится в околоцентромерном участке 2-ой хромосомы.
Симптомы
По тяжести клинического течения выделяют тяжелую, среднюю и легкую степени галактоземии.
Первые клинические признаки галактоземии тяжелой степени развиваются очень рано, в первые дни жизни ребенка. Вскоре после кормления новорожденного грудным молоком или молочной смесью возникает рвота и расстройство стула (водянистый понос), нарастает интоксикация. Ребенок становится вялым, отказывается от груди или бутылочки; у него быстро прогрессируют гипотрофия и кахексия. Ребенка могут беспокоить метеоризм, кишечные колики, обильное отхождение газов.
В процессе обследования ребенка с галактоземией неонатологом выявляется угасание рефлексов периода новорожденности. При галактоземии рано появляется стойкая желтуха различной степени выраженности и гепатомегалия, прогрессирует печеночная недостаточность. К 2-3 месяцу жизни возникают спленомегалия, цирроз печени, асцит.
Нарушения процессов свертывания крови приводит к появлению кровоизлияний на коже и слизистых оболочках. Дети рано начинают отставать в психомоторном развитии, однако степень интеллектуальных нарушений при галактоземии не достигает такой тяжести, как при фенилкетонурии. К 1-2 месяцам у детей с галактоземией выявляется двусторонняя катаракта. Поражение почек при галактоземии сопровождается глюкозурией, протеинурией, гипераминоацидурией. В терминальной фазе галактоземии ребенок погибает от глубокого истощения, тяжелой печеночной недостаточности и наслоения вторичных инфекций.
При галактоземии средней тяжести также отмечается рвота, желтуха, анемия, отставание в психомоторном развитии, гепатомегалия, катаракта, гипотрофия. Галактоземия легкой степени характеризуется отказом от груди, рвотой после приема молока, задержкой речевого развития, отставанием ребенка в массе и росте. Однако даже при легком течении галактоземии продукты обмена галактозы токсическим образом воздействуют на печень, приводя к ее хроническим заболеваниям.
Галактоземия может протекать в моносимптомной форме, при которой обнаруживается только поражение ЦНС, катаракта или диспепсические расстройства. Описан вариант бессимптомной (асимптоматической) галактоземии Дюарте, при которой недостаточность ферментов выявляется только при биохимическом исследовании крови.
Диагностика
Для снижения риска развития осложнений при галактоземии необходимо как можно более раннее выявление патологии. Возможна пренатальная диагностика галактоземии, включающая проведение биопсии хориона, амниоцентеза с последующим исследованием ворсин и амниотической жидкости.
В России, согласно современным стандартам, осуществляется скрининг новорожденных на следующие наследственные заболевания: фенилкетонурию, врожденный гипотиреоз, галактоземию, адрено-генитальный синдром и муковисцидоз. Неонатальный скрининг проводится на 3-5 сутки у доношенных детей и 7-10 сутки – у недоношенных. С этой целью производится забор капиллярной крови, которая переносится на фильтровальную бумагу и виде высушенных пятен отправляется в генетическую лабораторию.
Если при неонатальном скрининге у ребенка выявляется подозрение на галактоземию, проводится повторное решающее тестирование. В случае повторного обнаружения высокого уровня галактозы в крови или низкого уровня исследуемого фермента, ребенку устанавливается диагноз галактоземии. Сведения о таком ребенке сообщаются участковому педиатру, а семья новорожденного приглашается на консультацию генетика в медико-генетическую консультацию. Врач-генетик проводит подробный анализ родословной, выполняет генетическое тестирование для выявления мутантного гена, объясняет специфику питания ребенка с галактоземией.
Иногда для диагностики галактоземии прибегают к определению уровня галактозы в моче, проведению нагрузочных проб с галактозой и глюкозой. Мониторинг биохимических показателей крови и общего анализа крови и мочи при галактоземии позволяет определить степень повреждения внутренних органов (почек, печени и тд ).
Дети с галактоземией нуждаются в консультации детского невролога, детского офтальмолога, проведении электроэнцефалографии, УЗИ органов брюшной полости, биомикроскопии глаза. В некоторых случаях показана пункционная биопсия печени.
Галактоземию следует дифференцировать от других гликогенозов, сахарного диабета I типа, врожденной атрезии желчных протоков, гепатита, гемолитической болезни новорожденных.
Лечение
Основная роль в лечении галактоземии принадлежит диетотерапии. Особенность питания при галактоземии заключается в пожизненном исключении из рациона продуктов, содержащих лактозу и галактозу: любого молока (женского, коровьего, козьего, детских молочных смесей, низколактозных смесей и пр. ), всех молочных продуктов, хлеба, выпечки, колбас, конфет, маргаринов и тд При галактоземии запрещается употребление растительных и животных продуктов, содержащих потенциальные источники галактозы – галактозиды (бобовые, соя) и нуклеопротеины (почки, печень, яйца и тд ).
Дети, страдающие галактоземией, обеспечиваются специальными смесями на основе изолята соевого белка, гидролизата казеина, синтетических аминокислот, а также безлактозными казеинпредоминантными молочными смесями. С 4-х месячного возраста вводятся фруктовые и ягодные соки; с 4,5 месяцев – фруктовое пюре; с 5 месяцев – овощное пюре; с 5,5 месяцев – безмолочные каши из кукурузной, гречневой или рисовой муки в разведении специализированной смесью; с 6 месяцев – мясной прикорм на основе мяса кролика, цыпленка, индейки, говядины; с 8 месяцев – рыба. Альтернативным источником углеводов для пациентов с галактоземией служат продукты на основе фруктозы.
Для улучшения метаболических процессов назначаются поливитамины, кокарбоксилазу, АТФ, оротат калия. Лицам с галактоземией противопоказан прием спиртовых настоек и гомеопатических препаратов, поскольку последние содержат лактозу.
Дети с речевыми нарушениями нуждаются в консультации логопеда и целенаправленной работе по коррекции ОНР.
Прогноз
Лечение галактоземии, начатое с первых дней жизни позволяет избежать развития цирроза, катаракты, олигофрении. Если лечение начато в более поздние сроки, когда уже произошло поражение печени и ЦНС, с помощью рациональной диетотерапии прогрессирование заболевания можно замедлить. При тяжелых формах галактоземии может быть летальный исход.
Диспансерное наблюдение ребенка с галактоземией осуществляется педиатром, генетиком, диетологом, детским окулистом и детским неврологом. Детям с галактоземией присваивается инвалидность.
Профилактика
Учитывая наследственную обусловленность галактоземии, медико-генетическое консультирование рекомендуется пройти будущим родителям, в чьих семьях есть родственники или дети с данным заболеванием. Беременным с высоким риском рождения ребенка с галактоземией, следует ограничить употребление молочных продуктов.
Источник