
Фокальная дермальная гипоплазия синдром гольтца

а) Эпидемиология. Фокальная дермальная гипоплазия Гольтца (ФДГ), впервые описанная в 1934 г., получила свое название благодаря описанию Гольтцем и соавт. троих пациентов. В дальнейшем было описано более 200 зарегистрированных случаев болезни.
б) Этиология, патогенез, генетика. Фокальная дермальная гипоплазия представляет собой Х-сцепленное доминантное заболевание, которое обычно имеет летальное течение у пациентов мужского пола. Полагают, что клинические случаи с описанием больных мужского пола связаны с мозаицизмом соматических мутаций (этот факт подтвержден для синдрома недержания пигмента); выживание больных мальчиков обеспечивает наличие нормальных клеток.
Ген, в котором развивается мутация — PORCN, представляет собой человеческий гомолог гена porcupine у дрозофил. Считается, что PORCN важен для процесса пальмитоиляции и секреции белка Wnt, основного регулятора развития кожи и костей.
в) Клиника:
1. Кожные проявления. Основными диагностическими признаками при фокальной дермальной гипоплазии Гольтца (ФДГ) являются кожные изменения. К ним относятся линейная, точечная, полосатая решетчатая атрофии с телеангиэктазиями. Решетчатая атрофия представляет собой мелкие точечные вдавления в коже, которые располагаются вдоль линий Блашко.
Участки истонченной или отсутствующей кожи неравномерно распределены по поверхности тела, вследствие чего выпячивания жировой ткани имеют вид желто-розовых разрастаний над поверхностью кожи. Эти разрастания легко сдавливаются. Папилломы, которые могут быть мягкими или сосудистыми, развиваются на протяжении всей жизни и чаще поражают перигениталь-ные, периоральные, интертригинозные участки и слизистые оболочки.
К другим кожным симптомам относятся очаговая алопеция, ломкие или редкие волосы, а также гиперкератоз ладоней и ступней. У некоторых пациентов отмечался гипергидроз, еще у части пациентов была выявлена врожденная аплазия кожи.
2. Сочетанные системные проявления. В патологический процесс при фокальной дермальной гипоплазии Гольтца (ФДГ) также вовлекаются другие органы и системы, наиболее часто это костная, центральная нервная система, зубы и глаза. Типичным является поражение глаз в виде микрофтальма и колобомы, поэтому больным с диагнозом ФДГ необходимо проводить полное офтальмологическое обследование.
Также распространены олигодонтия, дисплазия зубов и дефекты зубной эмали. Перечень костных аномалий слишком велик, чтобы перечислять все эти симптомы; наиболее часто встречаются полосчатая остеопатия (образование вертикальных полосок на костях), синдактилия (как кожная, так и костная), асимметрия и низкорослость. Примерно в 15% случаев выявляется задержка умственного развития. В небольшом количестве случаев были описаны дефекты других органов и систем, в том числе — аномалии сердца, передней брюшной стенки и почек. Совершенно необязательно, что эти симптомы являются неотъемлемыми компонентами синдрома фокальной дермальной гипоплазии Гольтца (ФДГ).
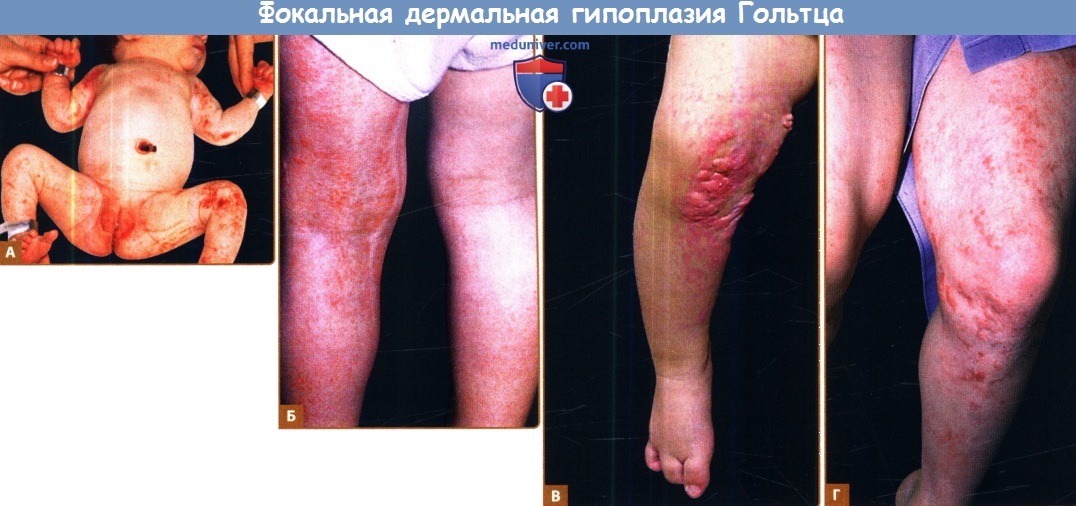
Фокальная дермальная гипоплазия Гольтца.
А. Полосчатое и очаговое поражение кожи новорожденного. Обратите внимание на сходность в распространении патологических элементов с синдромом недержания пигмента.
Б, В. Та же девочка в возрасте двух лет с атрофией и выпячиваниями жировой ткани.
Г. Молодой человек, у которого преобладают эритема и атрофия с сухостью и образованием чешуек.
г) Диагностика и дифференциация. Диагностика фокальной дермальной гипоплазии Гольтца (ФДГ) относительно проста. Решетчатая атрофия была описана также при Х-сцепленном доминантном синдроме Коранди-Хюнерманна (точечная хондродисплазия), в отличие от которого при фокальной дермальной гипоплазии Гольтца не встречается ихтиоз, и при котором, в отличие от фокальной дермальной гипоплазии Гольтца, не бывает выпячиваний подкожно-жировой клетчатки.
Полосчатое распространение атрофических участков, точно так же, как и другие системные проявления, могут напоминать синдром недержания пигмента, но характерные для недержания пигмента образование пузырей, гиперкератоз и гиперпигментация, при фокальной дермальной гипоплазии Гольтца не выявляются. При микрофтальме и линейных кожных дефектах с микрофтальмом, дермальной аплазией и склерокорнеа кожные проявления ограничены областью головы и шеи; отмечается атрофия и рубцевание кожи, более сходное с врожденной аплазией кожи, но не с дермальной атрофией. Заболевания имеют сходные глазные проявления.
д) Лечение и прогноз. Специфических методов лечения кожных и системных проявлений фокальной дермальной гипоплазии Гольтца нет. Папилломы могут удаляться, если они нарушают функции органа. Хороший косметический эффект может давать применение сосудистых лазеров для уменьшения эритемы в телеангиэктатических областях. Как и при большинстве Х-сцепленных доминантных заболеваний, степень клинических проявлений и тяжести течения болезни значительно варьирует.
Это затрудняет прогнозированрре заболевания в раннем грудном возрасте, поэтому обычно разумнее оставлять составление прогнозов на будущее, когда системные проявления станут более очевидными. Важны сведения о прерванных беременностях (измененное соотношение детей мужского пола к детям женского пола и повышенная частота прерванных беременностей позволяют предположить, что мать является носителем мутации). Оба родителя должны проходить тщательное обследование; у отцов могут отмечаться легкие симптомы болезни, предположительно они являются носителями постзиготных мутаций гена FDH.
– Рекомендуем далее ознакомиться со статьей “Признаки наследственных иммунодефицитов и их классификация”
Редактор: Искандер Милевски. Дата публикации: 25.12.2018
Источник
 Синдром, описанный Гольтцом в 1962 году, представляет многопорочное сочетание, последствие некоторых аномалий экто- и мезодермального развития, поражающих, в особенности, кожу, мышцы, скелет и глаза.
Синдром, описанный Гольтцом в 1962 году, представляет многопорочное сочетание, последствие некоторых аномалий экто- и мезодермального развития, поражающих, в особенности, кожу, мышцы, скелет и глаза.
Появляется у детей с рождения. До 1970 г. было опубликовано только 30 клинических наблюдений.
Этиопатогенез синдрома Горлина—Гольтца.
Этиология неизвестна. Генотипический характер синдрома вероятен, но не доказан. Кариотип—нормальный. Синдром появляется преимущественно у детей женского пола.
Синдром Горлина—Гольтца с клинической точки зрения похож на синдром Ротмунда, носящий выраженный семейный и наследственный характер, передающийся рецессивно-аутосомным образом и так же преимущественно появляющийся у женского пола.
Этот синдром известен в литературе под другими названиями, а именно:
- фокальная дермальная гипоплазия;
- синдром Гольтца-Петерсона-Горлина-Бавитца;
- синдром FDH;
Симптоматология синдрома Горлина—Гольтца
Кожные явления. У новорожденных кожные поражения могут проявляться в 3 клинических формах:
- Эритематозные, неправильной формы зоны, расположенные на туловище и на конечностях; позже эритематозные поражения становятся бледными и стертыми;
- Псевдобулезный аспект в некоторых небольших зонах кожи.
- Небольшие или более обширные зоны кожной врожденной аплазии, расположенные неравномерно, преимущественно на туловище и на конечностях.
Эти зоны могут чередоваться с некоторыми выпуклыми поражениями на поверхности кожи или даже с небольшими липоматозными опухолями. Со временем, зоны кожной аплазии превращаются в зоны с остаточными явлениями кожной атрофии или в зоны дермальной атрофии с липоматозом. В пораженных зонах появляется обычно гиперпигментация, однако, с одновременным наличием и зон гипопигментации.
У детей раннего возраста и дошкольника кожные поражения отличаются от новорожденного, проявляясь также в 3 клинических формах.
- Круглые или овальные зоны, с диаметром в несколько сантиметров, цвета охры или светло-желтого цвета из-за развивающегося липоматоза на здоровой кожной ткани, или в апластических кожных зонах, оставшихся после периода новорожденности. Чаще всего, они локализованы на туловище и конечностях.
- Участки дермальной гипоплазии, сочетающиеся с телеангиэктазией и с пигментными пятнами разных размеров (от нескольких мм до нескольких см), расположенными в виде зон, либо линейно в виде ленты.
- Ангиофиброматозный папилломатоз вокруг отверстий, локализованный, в особенности, на околоротовых и околоанусовых покровах или на слизистой миндалин и мягкого неба.
Кожные придатки сильно изменены. Отмечается: очаговая алопеция; ангидроз; искривление ногтей, хрупкость и в некоторых случаях даже их отсутствие.
Аномалии зубов: зубы небольших размеров, патологической формы или с порочной посадкой, иногда гипоплазия зубной эмали.
Аномалии скелета и мышц: синдактилии очень разнообразного типа и сочетания, проявляющиеся чаще всего только как кожные синдактилии; когда затронуты и кости, синдактилии появляются в особенности на III и IY пальцах обеих конечностей. В другом случае, синдактилии сочетаются с полидактилией или клинодактилией (в некоторых случаях последние представляют единственные аномалии пальцев). Гораздо реже отмечается аплазия или гиплоплазия разных пальцев (олигодактилия); сколиозы; genu valgum.
Глазные аномалии — разные (существуют приблизительно в 50% случаев), все ведут к тяжелым расстройствам зрения. Встречаются: микрофтальмия, часто в сочетании с микроцефалией; коллобома ириса и хороида; отсутствие ириса; подвывих хрусталика, косоглазие.
Патологическая анатомия синдрома Горлина—Гольтца.
Во всех случаях эпидермис не поражен. Характерные поражения локализованы в дерме. Структура дермы совершенно изменена, появляются гипопластические зоны, сопровождающиеся местными жировыми и ангиоматозными изменениями соединительной ткани: коллаген дермы пропитан интерстициальной жидкостью.
Течение и прогноз синдрома Горлина—Гольтца
Клиническое течение, благоприятное с точки зрения кожных проявлений; после выздоровления выявляются лишь малозначительные остаточные явления в дерме. Глазные признаки, без тенденции к регрессу, могут закончиться окончательным пороком.
Лечение синдрома Горлина—Гольтца.
При синдроме Гольтца не существует эффективного лечения, как и при всех других глазных и кожных синдромах, обусловленных аномалиями экто- и мезодермы. Генетический совет может оказаться полезным в жизни этих больных. Врач обязан информировать семью о наследственном характере аномалии и привлечь внимание родителей на возможность ее появления у наследников.
Источник
29.06.2010г.
 Синоним: синдром Гольтца. Заболевание описано в 1962 г. R. Goltz с соавт.
Синоним: синдром Гольтца. Заболевание описано в 1962 г. R. Goltz с соавт.
Минимальные диагностические признаки: участки истонченной кожи.
Клиническая характеристика
Кожные изменения характеризуются обширными сетчатыми или линейными участками истонченной кожи с выпячиванием жировой клетчатки (100%), полным отсутствием кожного покрова в некоторых участках тела, пигментными или депигментированными полосами, телеангиэктазиями, папилломами на губах, деснах, на основании языка, во влагалище. Папилломы также могут отмечаться в паховой, подмышечной и около пупочной областях.
Поражение кожи может проходить несколько стадий:воспалительную, десквамативную, пузырчатую, уртикарную или стадию выраженной эритемы. Отмечаются фолликулярный гиперкератоз, папулезные изменения, редкие ломкие волосы, отсутствие или дистрофия ногтей.
Наблюдаемые дефекты скелета: низкий рост, микрокрания, округлая форма черепа, заостренный подбородок, истончение и искривление носовой перегородки, прогнатизм, кифоз, сколиоз, слияние и сакрализация позвонков, скрытая расщелина позвоночника, рудиментарный хвост, асимметрия лица, туловища и конечностей, пороки развития дистальных отделов конечностей (гипоплазия или отсутствие пальцев, полигактилия, клешневидная кисть, синдактилия, слияние фаланг, камптодактилия, клинодактилия, вальгусная деформация), генерализованный остеопороз.
Наблюдаются анофтальмия, микрофтальмия, гипертелоризм, аниридия, нистагм, косоглазие, гетерохромия и колобома радужки, голубые склеры, подвывих хрусталика, колобома сосудистой оболочки, сетчатки и зрительного нерва, де- или гиперпигментация радужки, помутнение роговицы и стекловидного тела, эктропион века, птоз, закупорка слезных каналов.
Отмечаются недоразвитие нижней челюсти, аномалии прикуса, микродонтия, дисплазия и агенезия зубов, аномальное расположение зубов, дефекты эмали и кариес, расщелина губы, высокое арковидное небо, дефект альвеолярных отростков, срединная расщелина языка, двойная уздечка языка, гемигипоплазия языка, гипертрофия десен.
Имеют место недоразвитие крыльев носа, выступающие асимметричные ушные раковины, гипоплазия завитка, преаурикулярные выросты, смешанная глухота, шейные фистулы, гипоплазия I и V пальцев кисти, расхождение прямых мышц живота, омфалоцеле, паховая и пупочная грыжа, выпадение прямой кишки, асимметрия молочных желез с латеральным расположением ареол, пороки сердца (дефекты межпредсердной перегородки, стеноз легочного ствола), аномалии почек и мочеточников. Характерна умственная отсталость. Популяционная частота неизвестна.
Соотношение полов — М0:Ж1.
Тип наследования — Х-сцепленный доминантный.
Дифференциальный диагноз: врожденная пойкилодермия, синдром недержания пигмента, синдром эпидермальных невусов.
«Наследственные синдромы и медико-генетическое консультирование»,
С.И. Козлов, Е.С. Еманова
Читайте далее:
- Паллистера синдром (ulnarmammary syndrome, type Pallister)
- Плечелучевой синостоз (humeroradial synostosis)
- Прадера-Вилли cиндром (Prader-Willi syndrome)
- Расщелина губы или неба и сращение век (cleft lip or palate and filiform fusion of eyelids)
- Сетчатки дисплазия (retinal dysplasia)
- Сфероцитоз наследственный (spherocytosis)
- Тромбоцитопения с отсутствием лучевой кости (thrombocytopenia with absent radius)
- Фиброматоз десен, депигментация и микрофтальмия (gingival fibromatosis, depigmentation and microphthalmia)
- Хондродистрофия с полидактилией, тип Маевского (short ribpolydactyly syndrome, Majewski type)
- Хромосомы 13q- синдром (chromosome 13q- syndrome)
- Церебро-косто-мандибулярный синдром (cerebro-costo-mandibuear syndrome)
- Эктопия сердца (ectopia cordis)
- Паллистера-W синдром (Pallister-W syndrome)
- Поланда синдром (Poland syndrome)
- Прогерия (progeria)
- Расщелина еубы и неба, эктодермальная дисплазия и синдактилия (cleft lip-palate, ectodermal displasia and syndactyly)
- Синдактилия (syndactyly)
- Тапетохориоидальная дистрофия (tapetochorioidae dystrophy)
Источник
Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Эктомезодермальная очаговая дисплазия (син.: синдромы Гольтца, Гольтца-Горлина, фокальная дермальная гипоплазия, синдром мезоэктодермальной дисплазии) – редкое заболевание, вероятно, генетически неоднородное, но в большинстве случаев наследуемое Х-сцепленно доминантно с летальным исходом у плодов мужского пола, генный локус Хр22.31. Встречается почти исключительно у женщин.
[1], [2], [3], [4], [5], [6], [7], [8]
Патоморфология дисплазии эктомезодермальной очаговой
В атрофических участках кожи отмечается истончение дермы при нормальном строении эпидермиса. Подкожный жировой слой располагается близко к эпидермису, иногда на уровне поверхностного сосудистого сплетения. В сосочковом слое увеличено число сосудов, отмечается разреженность волокнистых структур. Коллагеновые волокна истончены, расположены в виде отдельных нитей. Придатков кожи мало, однако строение их в пределах нормы. Подобная картина напоминает поверхностный липоматозный невус кожи Гоффмана-Цуркалле. В участках гипоплазии с жировыми выпячиваниями выявляется аналогичная, но наиболее выраженная картина. В области папилломатозных разрастаний слизистых оболочек гистологическая картина соответствует папилломе с повышенной васкуляризациеи стромы. При электронно-микроскопическом исследовании атрофичных участков кожи обнаружены фибробласты с длинными цитоплазматическими выростами и слаборазвитой эндоплазматической сетью, вокруг которых располагаются тонкие фибриллы без поперечной исчерченности, возможно, комплексы тропоколлагена и проколлагена. Зрелые коллагеновые волокна по своей структуре не отличаются от нормальных. В подкожной основе преобладают жировые клетки с множеством жировых вакуолей – молодые формы, указывающие на пролиферацию жировых клеток.
Гистогенез кожных изменений при дисплазии эктомезодермальной очаговой связывают с нарушением кинетики роста фибробластов при их нормальной синтетической активности, следствием чего является скудность соединительнотканных элементов дермы. Одновременно происходит пролиферация клеток подкожной клетчатки, замещающих неполноценную дерму.
Симптомы
Клинически проявляется атрофопойкилодермическими изменениями кожи с грыжевидными выпячиваниями подкожной клетчатки, аномалиями скелета, зубов и глаз, появляющихся с рождения. Часто наблюдаются папилломы на слизистых оболочках губ, влагалища, заднего прохода, поражение придатков кожи в виде дистрофии ногтей, нарушения роста волос с гнездным облысением и потоотделения.
Отмечается умственное недоразвитие с эпилептиформными припадками.
Источник
Детский врач

- Вы студент медик? Интерн? Детский врач? Добавьте наш сайт в социальные сети!
Синдром, описанный Гольтцом в 1962 году, представляет многопорочное сочетание, последствие некоторых аномалий экто- и мезодермального развития, поражающих, в особенности, кожу, мышцы, скелет и глаза.
Появляется у детей с рождения. До 1970 г. было опубликовано только 30 клинических наблюдений.
Этиопатогенез синдрома Горлина—Гольтца.
Этиология неизвестна. Генотипический характер синдрома вероятен, но не доказан. Кариотип—нормальный. Синдром появляется преимущественно у детей женского пола.
Синдром Горлина—Гольтца с клинической точки зрения похож на синдром Ротмунда, носящий выраженный семейный и наследственный характер, передающийся рецессивно-аутосомным образом и так же преимущественно появляющийся у женского пола.
Этот синдром известен в литературе под другими названиями, а именно:
- фокальная дермальная гипоплазия;
- синдром Гольтца-Петерсона-Горлина-Бавитца;
- синдром FDH;
Симптоматология синдрома Горлина—Гольтца
Кожные явления. У новорожденных кожные поражения могут проявляться в 3 клинических формах:
- Эритематозные, неправильной формы зоны, расположенные на туловище и на конечностях; позже эритематозные поражения становятся бледными и стертыми;
- Псевдобулезный аспект в некоторых небольших зонах кожи.
- Небольшие или более обширные зоны кожной врожденной аплазии, расположенные неравномерно, преимущественно на туловище и на конечностях.
Эти зоны могут чередоваться с некоторыми выпуклыми поражениями на поверхности кожи или даже с небольшими липоматозными опухолями. Со временем, зоны кожной аплазии превращаются в зоны с остаточными явлениями кожной атрофии или в зоны дермальной атрофии с липоматозом. В пораженных зонах появляется обычно гиперпигментация, однако, с одновременным наличием и зон гипопигментации.
У детей раннего возраста и дошкольника кожные поражения отличаются от новорожденного, проявляясь также в 3 клинических формах.
- Круглые или овальные зоны, с диаметром в несколько сантиметров, цвета охры или светло-желтого цвета из-за развивающегося липоматоза на здоровой кожной ткани, или в апластических кожных зонах, оставшихся после периода новорожденности. Чаще всего, они локализованы на туловище и конечностях.
- Участки дермальной гипоплазии, сочетающиеся с телеангиэктазией и с пигментными пятнами разных размеров (от нескольких мм до нескольких см), расположенными в виде зон, либо линейно в виде ленты.
- Ангиофиброматозный папилломатоз вокруг отверстий, локализованный, в особенности, на околоротовых и околоанусовых покровах или на слизистой миндалин и мягкого неба.
Кожные придатки сильно изменены. Отмечается: очаговая алопеция; ангидроз; искривление ногтей, хрупкость и в некоторых случаях даже их отсутствие.
Аномалии зубов: зубы небольших размеров, патологической формы или с порочной посадкой, иногда гипоплазия зубной эмали.
Аномалии скелета и мышц: синдактилии очень разнообразного типа и сочетания, проявляющиеся чаще всего только как кожные синдактилии; когда затронуты и кости, синдактилии появляются в особенности на III и IY пальцах обеих конечностей.
В другом случае, синдактилии сочетаются с полидактилией или клинодактилией (в некоторых случаях последние представляют единственные аномалии пальцев).
Гораздо реже отмечается аплазия или гиплоплазия разных пальцев (олигодактилия); сколиозы; genu valgum.
Глазные аномалии — разные (существуют приблизительно в 50% случаев), все ведут к тяжелым расстройствам зрения. Встречаются: микрофтальмия, часто в сочетании с микроцефалией; коллобома ириса и хороида; отсутствие ириса; подвывих хрусталика, косоглазие.
Патологическая анатомия синдрома Горлина—Гольтца.
Во всех случаях эпидермис не поражен. Характерные поражения локализованы в дерме. Структура дермы совершенно изменена, появляются гипопластические зоны, сопровождающиеся местными жировыми и ангиоматозными изменениями соединительной ткани: коллаген дермы пропитан интерстициальной жидкостью.
Течение и прогноз синдрома Горлина—Гольтца
Клиническое течение, благоприятное с точки зрения кожных проявлений; после выздоровления выявляются лишь малозначительные остаточные явления в дерме. Глазные признаки, без тенденции к регрессу, могут закончиться окончательным пороком.
Лечение синдрома Горлина—Гольтца.
При синдроме Гольтца не существует эффективного лечения, как и при всех других глазных и кожных синдромах, обусловленных аномалиями экто- и мезодермы. Генетический совет может оказаться полезным в жизни этих больных. Врач обязан информировать семью о наследственном характере аномалии и привлечь внимание родителей на возможность ее появления у наследников.
- Вы студент медик? Интерн? Детский врач? Добавьте наш сайт в социальные сети!
Источник: https://detvrach.com/facultet/dermatovenerologia/sindrom-gorlina-golttsa/
Синдром Горлина-Гольца * My dermatology

Синдром Горлина-Гольца
Это заболевание наследуется аутосомно-доминантно. Онопоражает кожу (первично-множественный базальноклеточный рак, мелкие углубленияна ладонях и подошвах), скелет (одонтогенные кисты нижней челюсти, кифосколиози др.), а также другие органы — мягкие ткани, глаза, ЦНС, железы внутреннейсекреции.
Синонимы: синдром Гольтца—Горлина,невобазоцеллюлярный синдром, синдром ба-зальноклеточных невусов, синдромневоидной клеточной эпителиомы.
Эпидемиология
Частота
Синдром Горлина—Гольца — отнюдь не редкость, однако точныхданных нет.
Возраст
Почти все изменения — врожденные, базальноклеточный ракможет появиться в позднем детском возрасте.
Наследственность
Тип наследования — аутосомно-доминант-ный, с различнойпенетрантностью.
Раса
Большинство больных — белые, однако заболевание встречаетсяи среди представителей цветных рас.
Пол
Мужчины и женщины болеют одинаково часто.
Провоцирующие факторы
Инсоляция: базальноклеточный рак чаще встречается наоткрытых участках тела.
Анамнез
Течение
Первые опухоли появляются в детском или подростковомвозрасте и затем продолжают возникать в течение всей жизни.
Общее состояние
Аномалии развития — крипторхизм; гидроцефалия; колобома,катаракта и глаукома, приводящие к слепоте.
Семейный анамнез
Отягощен.
Физикальное исследование
Внешний вид
Характерное лицо с выступающими лобными буграми, широкойпереносицей и широко расставленными глазами. Количество опухолей можетдостигать нескольких сотен.
Базальноклеточный рак кожи Элементы сыпи. Полупрозрачныепапулы или узлы, нередко изъязвленные, диаметром от 1 до 10 см,гиперпигментированные или не отличающиеся по цвету от окружающей кожи.Базальноклеточный рак при синдроме Горлина—Гольца редко бывает инвазивным. Опухолина веках, шее и в подмышечных впадинах могут иметь ножку. Расположение.Двустороннее, часто симметричное.
Локализация. Лицо (рис. 10-10), шея, верхняя часть туловища,подмышечные впадины. Конечности и волосистая часть головы почти никогда непоражаются.
Углубления на ладонях и подошвах
У половины больных — точечные углубления, достигающиенескольких миллиметров в диаметре и 1 мм в глубину (рис. 10-11). На дне почтивсегда обнаруживают телеангиэк-тазии.
Количество элементов достигает несколькихсотен; больше всего углублений на боковых поверхностях ладоней, подошв ипальцев рук. Эти высыпания возникают из-за преждевременного слущивания роговогослоя эпидермиса.
Изредка они представляют собой базальноклеточный рак.
Другие органы
Кости. Множественные одонтогенные кисты нижней челюсти,односторонние или двусторонние. Неправильное расположение зубов, расщеплениеребер, воронкообразная грудная клетка, укорочение IV пястных костей, сколиоз, кифоз.
Глаза. Косоглазие, гипертелоризм, дистопия внутренних угловглазной щели, врожденная слепота.
ЦНС. Агенезия мозолистого тела, медулло-бластома,пластинчатое обызвествление серпа большого мозга. Изредка — умственнаяотсталость.
Новообразования внутренних органов. Фибро-саркомы челюстей;фибромы, тератомы и цистаденомы яичников.
Рисунок 10-10. Синдром Горлина—Гольца: базальноклеточныйрак. Множественные узлы на правой стороне лица и большой рубец на правойщеке, оставшийся после иссечения одонтогенной кисты
Дополнительные исследования
Патоморфология кожи
Все формы базальноклеточного рака: узел-ково-язвенная,поверхностная, склеродер-моподобная, фиброэпителиальная, кистоз-ная.
Лучевая диагностика
Пластинчатое обызвествление серпа большого мозга.
Диагноз
Достаточно клинической картины (углубления на ладонях иподошвах, характерные черты лица). Диагноз подтверждают с помощью биопсии.Заболевание нередко выявляют стоматологи и челюстно-лицевые хирурги, к которымбольные обращаются по поводу кист челюстей.
Этиология и патогенез
Локализация мутантного гена — 9q22.
Клиническое значение
Множественные злокачественные опухоли кожи требуют и отбольного, и от врача постоянной настороженности. Многократные операции могутпривести к образованию грубых рубцов.
Течение и прогноз
Новые опухоли появляются в течение всей жизни, поэтому забольным устанавливают диспансерное наблюдение. Своевременное выявление иоперативное лечение базальноклеточного рака помогает избежать образованиярубцов, уродующих больного.
Лечение
Опухоли, расположенные в центральной части лица, на ушныхраковинах и в их окружности, подлежат иссечению по методу Моса — синтраоперационной микроскопией замороженных горизонтальных срезов дляопределения объема операции. Для мелких опухолей на туловище и конечностяхиспользуют электрокоагуляцию или комбинированное местное лечение трети-ноином ифторурацилом (5% крем) в течение 25—30 сут.
Рисунок 10-11. Синдром Горлина—Гольца: углубления наладонях. Ладонь испещрена четко очерченными красноватыми углублениямиразмером 1—2 мм
Источник: https://dermatology.my1.ru/publ/fitzpatrick/1/sindrom_gorlina_golca/55-1-0-144
Базально-клеточный рак

Синонимы: базалиома, базально-клеточная эпи- телиома.
Определение
БКР является наиболее распространенным злокачественным новообразованием. Он встречается в 10 раз чаще, чем ПКР, и характеризуется медленным, но инфильтрирующим и деструктивным ростом. Поскольку мстастазирования практически не происходит, новообразование часто называют полузлокачественным.
Эпидемиология
Болеют преимущественно люди с кожей I и II фототипов. Пик заболеваемости приходится на период между 60-м и 80-м годами жизни.
Если БКР развивается на 2-3-м десятилетии жизни, это может свидетельствовать о наследственной предрасположенности в форме синдрома Горлина-Гольца.
Заболеваемость в Европе и США растет и составляет 100-200 случаев на 100000 населения в год, а в Австралии она в 10 раз выше.
Патогенез
Развитие БКР (не только при синдроме Горлина-Гольца) связано с нарушением регуляции сигнального каскада, запускаемого белковым фактором БНЬ.
Существует определенная связь с волосяным фолликулом и совпадение признаков с новообразованиями волосяного фолликула (трихобластным раком).
БКР рассматривается в настоящее время как самостоятельное новообразование без установленного наличия аденомы или предракового очага. Важнейшими эндогенными нарушениями являются следующие:
• Генетика — I и II фототипы кожи, при синдроме Горлина-Гольца — мутации в гене РТСН.
• Нарушения в сигнальном каскаде «Hedgehog- втооШепесНЗи», который регулируется ОН, РТСН, ТФР-р и ВСЬ2. Трансмембранный белок РТСН тормозит активацию этого сигнального пути основным его трансдуктором — трансмембранным белком «БтооФепес!», в результате чего активируется группа факторов транскрипции вИ, что практически неизбежно приводит к развитию БКР.
Важнейшими экзогенными факторами являются следующие:
• УФ-облучение типов В и А.
• Иммуносупрессия — ятрогенная супрессия Т-клеточного иммунитета после трансплантации органов.
• Ионизирующее излучение — лучевая терапия с латентным периодом свыше 20 лет при нагрузке кожи начиная примерно с 10 Гр.
• Канцерогены — мышьяк.
Клиническая картина
Примерно в 80% случаев БКР локализуется на лице, из них 90% — на носу, лбу, висках и щеках. На туловище часто наблюдаются множественные, поверхностно растущие, так называемые мультицентрические базалиомы, в то время как поражение кистей отмечается редко.
Чаще встречается поражение голени в форме язвы. БКР растет сравнительно медленно. Опасность состоит в неограниченной инфильтрации во все органы. Клинический спектр довольно широк.
Различают три основные клинические формы (узловатую, склеродермоподобную, поверхностную, или мультицснтричсскую) и другие особые формы. Узловой, или солидный, БКР и его варианты.
Эта форма встречается наиболее часто и представляет собой чаще всего блестящий узел на широком основании с телеангиэктазиями и валиком, приподнятым по краю по типу «нитки жемчуга». Иногда отмечается небольшое изъязвление в центре очага с коркой. Часто встречаются следующие варианты:
• кистозный БКР: новообразование выглядит прозрачным вследствие наличия кистозных участков. Дифференциальный диагноз: ги- дрокистома.
• пигментированный БКР: «жемчужины», имеющие цвет от коричневатого до серо-черного. Дифференциальный диагноз: меланома.
Склеродермоподобный БКР. Эта форма БКР имеет вид слегка вдавленного уплотненного рубца цвета кожи, поэтому часто ее ошибочно принимают за рубец. Приподнятый край по типу «нитки жемчуга» и телеангиэктазии могут отсутствовать.
Эту форму БКР трудно диагностировать и оперировать, так как распространение опухоли в длину и ширину невозможно оценить клинически. Часто она распространяется дальше макроскопически видимых границ (феномен «айсберга»). При ее выявлении следует срочно выполнить операцию с микроскопическим контролем. Поверхностный БКР.
Этот вид рака называют также базалиомой кожи туловища, поверхностным мультицентрическим или экзематозным БКР.
Он представляет собой плоскую, краснокоричневую, часто слегка шелушащуюся, незначительно приподнятую бляшку, которая по краю окружена мельчайшими, едва заметными, нитевидно расположенными узелками, иногда с единичными телеангиэктазиями. На туловище очаги часто множественные. Собирают анамнез о приеме препаратов мышьяка и наличии имму- носупрсссии.
Фиброэпителиома (опухоль Пинкуса). Смешанная, частично эпителиальная и частично мезенхимальная опухоль. Соответствует БКР со значительным преобладанием мезенхимальных компонентов. Часто расположена в нижней части туловища, напоминает фибромы. Изъязвленный БКР.
Поскольку эта форма опухоли безболезненна, пациенты «терпят» ее в течение многих лет. С ростом опухоли начинается деструкция тканей в центре очага с возникновением «разъедающей язвы» или распространение процесса деструкции вглубь тканей с возникновением «проникающей язвы».
При этой форме БКР процесс деструкции может распространяться через нос в основание и свод черепа, сосуды или позвоночник.
Поскольку заболевание причиняет сильные страдания, а удаление новообразования, как правило, не представляет проблем, хирургическое лечение всегда показано даже очень пожилым пациентам с многочисленными сопутствующими заболеваниями или деменцией.
При отсутствии лечения эта форма БКР может трансформироваться в ПКР с а1рессивным ростом.
Гистопатология
Базалоидная опухоль с тяжами базалоидных клеток, идущих с нижней стороны эпидермиса, с палисадным расположением внешних клеточных рядов, часто с гнездными скоплениями. Дсрмально расположенные опухолевые скопления и тяжи, инфильтрирующие в строму при склеродермоподобной форме и опухоли Пин- куса вариабельное лимфо