
Франческетти синдром код мкб

Синдром Франческетти или Тричера Коллинза – врождённое заболевание, которое встречается крайне редко. Согласно статистике, эта патология наблюдается у одного из пятидесяти тысяч новорождённых. Стоит отметить, что существуют несколько стадий развития болезни. Если говорить о начальной, то здесь ребёнку практически ничего не угрожает. У него есть некоторые дефекты внешне, но это никак не отображается на умственном развитии малыша.
Если заболевание протекает без осложнений, то дети с таким синдромом могут вести активный образ жизни. Однако не всё так радужно. В случае тяжёлой стадии развития болезни ребёнок очень сильно страдает, у него практически отсутствует лицо. Это полностью исключает новорождённого из социума. В данной статье мы рассмотрим причины появления синдрома Франческетти, симптомы и методы лечения.
Что это такое?
Как уже было отмечено, синдром Коллинза является исключительно генетическим заболеванием. Его невозможно приобрести в течение жизни, он проявляется с рождения. Данная болезнь – один из подвидов дизостоза, который, в свою очередь, означает нарушение или неправильное развитие костных тканей. Если говорить о синдроме Франческетти, то здесь происходит деформация костей черепа, что не может не оказывать влияние на физическое и моральное состояние человека.

У новорождённого сразу заложены клинические проявления, от количества которых зависит тяжесть развития заболевания. Стоит отметить, что болезнь характеризуется нарушением в строении черепа, которое отображается на лице человека. Чаще всего страдают от синдрома дети, у родителей которых наблюдаются спонтанные мутации генов.
Причины появления болезни
Синдром является врождённым заболеванием, что исключает влияние каких-либо внешних и внутренних факторов на его развитие. Необходимо отметить, что недуг изначально заложен в аминокислотный код ребёнка, причём ещё до его появления на свет. Учёные сумели доказать появление разного рода мутаций генов в пятой хромосоме. Это не случайность, ведь именно она отвечает в организме человека за качество материала для скелета. Кроме того, говоря о строении хромосомы, можно отметить её размер: сама длинная структура в геноме.

Сами по себе мутации происходят на основе нарушения во внутриклеточном синтезе белка. Затем у человека наблюдают гаплонедостаточность. Другими словами, в организме просто не хватает белка для формирования лицевой части черепа. Основной причиной появления синдрома Франческетти является наследственность генов. Однако заболевание может развиться из-за новых мутаций, которые вызываются некоторыми факторами:
- радиоактивное излучение;
- токсоплазмоз;
- приём психотропных медикаментов.
Признаки синдрома
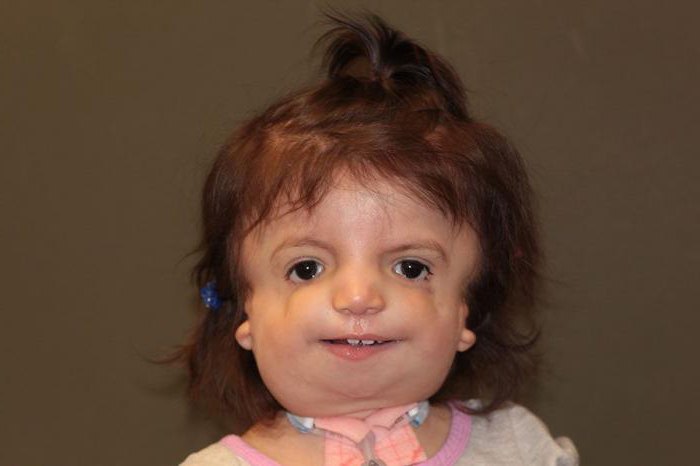
Так как данная болезнь характеризуется тем, что поражает ещё не родившийся плод, то первые симптомы можно обнаружить сразу после появления на свет. Более того, практически все признаки недуга проявляются сразу. Даже несведущий человек в медицине при мимолётном взгляде на больного может выявить определённые симптомы. Главным из них является нарушение формы глаз. Видно сразу человека с синдромом Франческетти – фото, представленные в статье, это наглядно демонстрируют.

Кроме вышеуказанного признака, есть ещё несколько, которые характеризуют данное заболевание. Стоит отметить, что все симптомы довольно ярко выражены, и их сложно не заметить. Итак, среди них наиболее распространёнными являются:
- сбой в строении скул и нижней челюсти;
- проблемы со слухом;
- нарушение в структуре подбородка;
- отсутствие ушных раковин;
- неправильный прикус.
Уровни развития недуга
В начале статьи было указано, что существует несколько стадий болезни, о которых пришло время поговорить подробнее. Синдром Франческетти (Коллинза) имеет три уровня развития. Первый и наиболее безопасный характеризуется небольшой гипоплазией костей лицевой части. Однако даже в этом случае человек меняется внешне, и самые внимательные могут заподозрить наличие заболевания.
Когда наступает вторая стадия, то здесь уже добавляются проблемы со слухом, неправильная форма глазных щелей, деформация нижней челюсти. В этом случае недуг становится более явным, и человек с синдромом внешне значительно отличается от обычного индивида.
Что касается последней, наиболее тяжёлой стадии, то тут имеет место полное отсутствие лица. На самом деле это очень страшно, ведь пациент просто автоматически исключается из общества, и с очень низкой долей вероятности что-то изменится. Кроме того, эти люди с годами страдают сильнее, ведь патология только усугубляется. Могут появиться признаки более тяжёлых болезней. Синдром Франческетти – сам по себе довольно опасный недуг, а с годами могут приобретаться заболевания и похуже.
Наиболее серьёзные осложнения
Помимо того что синдром и так ограничивает многие возможности человека, есть ещё и особые случаи, которые оказывают сильнейшее влияние на здоровье организма. Одним из наиболее тяжёлых осложнений принято считать деформацию ротового аппарата. Это связано с проблемами в строении зубов, что приводит к тому, что больной не может самостоятельно кушать.

Также могут возникнуть дополнительные сложности с дыханием, так как при синдроме Франческетти язык увеличивается в размерах и зарастают носовые проходы. Безусловно, эти факторы оказывают негативное влияние на дыхательную систему человека.
Диагностика
Стоит отметить, что выявление данного недуга происходит во время беременности. У сформировавшегося плода врачи диагностируют наличие синдрома Франческетти. Рекомендуется проводить исследование лицевой части с помощью биопсии. Однако этот способ не является безопасным, и поэтому врачи часто используют для этих целей УЗИ. Также необходимо у всех членов семьи взять анализы крови. После этого беременной женщине следует пройти фетоскопию. Заключительным этапом диагностики является взятие крови из плацентарных сосудов.
Причины синдрома Франческетти кроются в мутации генов, поэтому в зависимости от выраженности болезни врачи принимают решение о целесообразности дальнейших исследований. Если заболевание характеризуется экспрессивностью, то здесь сомнений быть не может. Однако в случае обнаружения некоторых отдельных симптомов специалисты обязаны продолжить наблюдение. Список мероприятий для выявления синдрома составляется врачом индивидуально, и в большинстве случаев это приносит свои плоды. Данное заболевание является довольно тяжёлым, и поэтому очень важно диагностировать его заранее, чтобы оставался шанс на исправление положения вещей.
Лечение синдрома Франческетти
К сожалению, необходимо констатировать тот факт, что в настоящее время медицина не готова предложить терапевтические методы лечения данной проблемы, их попросту не существует. Вся терапия направлена исключительно на паллиативную помощь. Человек, который отличается от всех остальных, сильно желает быть кому-то нужным. Поэтому в этом случае ценнее простой человеческой заботы трудно что-либо найти.

Если наблюдаются тяжёлые стадии заболевания, то следует делать операцию. Если у человека при этом есть проблемы со слухом, врачи рекомендуют использовать слуховой аппарат. Конечно, от моральной поддержки зависит очень многое. Сможет ли пациент поверить в себя? Сможет ли быть сильнее всех проблем? Довольно трудные вопросы. Это говорит о том, что психологическая помощь не может быть лишней никогда. В любой ситуации при возможности необходимо поддержать добрым словом. Вам это ничего не стоит, а больному это может открыть второе дыхание.
Прогноз
Стоит отметить, что в большинстве случаев люди с диагнозом синдром Франческетти проживают полноценную жизнь, находят супругов, рожают детей. Однако, что касается общества, то тут возникают определённые сложности. Социально адаптироваться таким людям нереально трудно, что, безусловно, негативно отражается на их здоровье.

Но если у человека не самая тяжёлая стадия болезни и он морально справился с давлением общественности, то шанс на полноценную жизнь возрастает. Главное – преодолеть проблему психологически и продолжать делать своё дело.
Заключение
Синдром Коллинза встречается довольно редко, однако является серьёзным заболеванием. Причиной данной болезни является наследственность, спонтанная мутация генов. Врачи во время обследований беременной женщины начинают проверку на наличие деформации лицевой части плода. Если исследование показало, что болезнь существует, специалист обязан составить курс терапии, который максимально облегчит жизнь человеку.
Источник
Синдром Тричера Коллинза – это генетическая (иногда наследственная) болезнь, сопровождающаяся деформациями костей и мягких тканей лица. К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, вырезки ткани век (колобомы), уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина или арковидная форма неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица. Диагноз устанавливается по данным клинического осмотра, биогенетического теста и семейного анамнеза. Лечение симптоматическое, направлено на улучшение слуха, устранение жизнеугрожающих деформаций и косметических дефектов хирургическим способом.
Общие сведения
У синдрома Тричера Коллинза есть несколько синонимов: челюстно-лицевой дизостоз, синдром Тричера Коллинза-Франческетти, мандибулофациальный дизостоз. Впервые патологию описал офтальмолог из Великобритании Эдвард Тричер Коллинз в 1900 году, поэтому наиболее распространено название, соответствующее его имени. Обширный обзор заболевания был сделан в 1949 году европейскими исследователями Э. Франческетти и Д. Клейном. В настоящее время понятие «синдром Тричера Коллинза» более распространено в Великобритании и США, а термин «синдром Франческетти-Клейна» чаще используется в странах Европы. Эпидемиология болезни составляет 1:50 000. Среди мальчиков и девочек заболеваемость одинакова.

Синдром Тричера-Коллинза
Причины
Развитие синдрома в 78-93% случаев обусловлено мутациями гена TCOF1, расположенного на пятой хромосоме в регионе 5q32. Данный ген кодирует производство ядерного фосфопротеина Treacle. У 7-9% пациентов причиной заболевания является дефект гена POLR1C, локализованного на шестой хромосоме, или гена POLR1D, находящегося на тринадцатой хромосоме. Они ответственны за синтез I и III РНК-полимеразы.
При мутациях в гене TCOF1 тип наследования синдрома аутосомно-доминантный с показателем пенетрантности 90%. Это означает, что при мутации в одной хромосоме из пары вероятность проявления болезни очень высока. У больного родителя риск рождения ребенка с синдромом Тричера Коллинза составляет 50%. Возможна наследственная передача дефекта и спорадические генетические изменения (новые мутации). Экспрессивность мутации переменная – в пределах одной семьи вероятно как ослабление, так и усиление симптомов заболевания у последующих поколений. При дефектах генов POLR1C и POLR1D наследование происходит по аутосомно-рецессивному типу. В парах, где родитель имеет синдром, вероятность рождения больного малыша составляет 25%.
Патогенез
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle. Данный протеин экспрессируется в большинстве тканей организма в эмбриональном и постэмбриональном периоде, участвует в переносе генетической информации с ДНК на РНК.
В основе синдрома чаще всего лежит нонсенс-мутация, приводящая к образованию преждевременного кодона терминации и развитию гаплонедостаточности – дефицита белка, необходимого для нормального формирования лицевой части черепа. Здоровый ген обеспечивает организм белком Treacle наполовину, но такого количества недостаточно для правильного развития лицевых структур. При изменениях в генах POLR1D и POLR1C процесс транскрипции ДНК нарушается из-за недостаточности фермента-катализатора ДНК-зависимой РНК-полимеразы. Клинические проявления синдрома такие же, как и при первичной недостаточности Treacle-протеина.
Симптомы
У больных наблюдаются аномалии в строении лица. Распространенным признаком, встречающимся в 80% случаев, является двусторонняя симметричная гипоплазия скуловых костей, инфраорбитального края и нижней челюсти. Внешне это проявляется своеобразным уплощенным бесформенным лицом, на котором выделяется нос, а остальные части «утоплены» в мягких тканях. Деформация челюсти обуславливает нарушение прикуса, формирование ортогнатии (постоянно приоткрытого рта). 89% больных имеют ограниченную возможность открывания рта и антимонголоидный тип разреза глаз с заметным опущением внешнего уголка. Данные особенности частично обусловлены патологическим строением височно-нижнечелюстного сустава.
У 69% пациентов определяется колобома радужки и нижних век в промежутке между средней и внешней третью, чаще она имеет треугольную форму. Ресницы на внешнем крае нижнего века отсутствуют. Небо арковидной формы, иногда сформирована расщелина (у 28% больных). Аномалии наружного уха представлены недоразвитием или полным отсутствием ушной раковины (микротией, анотией), атрезией наружного слухового прохода и деформацией слуховых косточек. Зачастую пациенты имеют кондуктивную тугоухость. В редких случаях диагностируется энхондрома, предкозелковые фистулы, аномальное строение сердца и позвоночника.
Осложнения
Микрогнатия и стеноз верхних дыхательных путей уже в первые годы жизни могут спровоцировать проблемы при приеме пищи и трудности дыхания вплоть до удушья. Своевременная диагностика заболевания позволяет спрогнозировать эти осложнения и предпринять меры по их предупреждению. Как правило, пациенты не имеют врожденных интеллектуальных расстройств, но при отсутствии коррекции нарушений слуха становится невозможным правильное формирование речи и обучение в обычных условиях. Дети начинают отставать в умственном развитии от сверстников, имеют задержку психического развития различной степени выраженности. В связи с наличием дефектов внешности и негативным отношением окружающих больные всех возрастов относятся к группе риска по возникновению депрессии, ипохондрии, тревожности и иных невротических расстройств.
Диагностика
Диагноз может быть установлен во время беременности или сразу после рождения. Обследование показано женщинам из группы риска и детям с врожденными лицевыми деформациями. В процессе диагностики принимают участие врачи-генетики и педиатры. Синдром Тричера-Коллинза необходимо дифференцировать с другими генетическими заболеваниями, при которых существует деформация лицевой части черепа, например, с синдромом Нагера и синдромом Гольденхара. Используются следующие методы:
- Осмотр, сбор анамнеза. Определяются характерные черепно-лицевые аномалии: недоразвитость костей скул и челюсти, деформация и гипоплазия ушных раковин, антимонголоидный тип глазных щелей, нарушения слуха и дефект верхнего неба. Иногда подтвержденный диагноз синдрома имеется у одного из родителей.
- Биогенетический тест. Антенатальное исследование включает молекулярный анализ образца ворсин хориона на 10-11 неделе беременности, фетоскопию и анализ крови из сосудов плаценты на 18-20 неделе. После родов выполняется забор крови из вены ребенка. В обоих случаях исследуется ген TCOF1. Заболевание подтверждается при наличии в нем мутации любого типа.
- Дородовое УЗИ. С 20-24 недели беременности ультразвуковое исследование плода способно выявить типичные изменения лица. Наиболее четко заметна двусторонняя аномалия ушей, гипоплазия скул и челюсти.
Дополнительно назначаются обследования, позволяющие своевременно обнаружить жизнеугрожающие состояния, оценить степень деформации костей черепа. Определяется эффективность кормления ребенка, уровень насыщения гемоглобина кислородом, ритмичность и глубина дыхания. Для диагностики сохранности слуха на 5-6 день жизни проводится аудиологическое тестирование. Инструментальная диагностика включает рентгенографию черепа, КТ и МРТ головного мозга.
Лечение синдрома
Специфической терапии не существует. Лечение нацелено на устранение симптомов и последствий заболевания, предполагает проведение хирургических операций и реабилитационных мероприятий. Объем процедур и сроки их выполнения устанавливаются индивидуально с учетом наличия угрозы для жизни больного, противопоказаний и рисков, связанных с оперативным вмешательством. Общая схема лечения включает:
- Восстановление глотания и дыхания. При развитии респираторного дистресс-синдрома осуществляется трахеостомия, дистракция подвижной челюсти, неинвазивная вентиляция легких. При невозможности потребления пищи устанавливается гастростома.
- Восстановление слуха. Деформация наружного и среднего уха устраняется хирургическим путем, но потеря слуха чаще обусловлена повреждением слуховых мелких косточек, поэтому оперативные вмешательства с целью устранения тугоухости неэффективны. Предпочтительна реабилитация слуховыми аппаратами.
- Устранение внешних дефектов. Деформации корректируются методами пластической и нижнечелюстно-лицевой хирургии. Применяется липоскульптурирование, хирургическая дистракция костей, установка трансплантатов и хирургическое восстановление неба.
Прогноз и профилактика
Комплексное лечение и реабилитация значительно улучшают качество жизни больных. При легкой и умеренной выраженности синдрома прогноз благоприятный. Профилактика затруднена, поскольку заболевание является генетическим, а мутации способны возникать спонтанно. Супружеским парам, в которых один родитель болен, необходимо медико-генетическое консультирование и перинатальная диагностика синдрома на ранних сроках беременности. Для снижения риска вынашивания больного ребенка рекомендуется процедура экстракорпорального оплодотворения с предварительным отбором генетически здоровых эмбрионов.
Источник
Синдром Франческетти является аутосомно-доминантным расстройством черепно-лицевого развития с переменной экспрессией. Широко известен как синдром Тришера Коллинза (TCS).
Назван в честь английского офтальмолога Э. Т. Коллинза, который в 1900 году описал основные компоненты этого состояния. Расстройство влияет на оба пола одинаково.
В Англии и на американском континенте эта аномалия описывается как «синдром Тришера Коллинза», на европейском континенте – «Мандибуло-лицевой дизостос» или «синдром Франческетти»
Другие названия:
- Синдром Франческетти-Звален-Клейн;
- Мандибулофациальный дистоз (MFD1);
- Синдром Тришера Коллинза-Франческетти;
- Зигоуроандибулярная дисплазия.

Характеристики
Синдром Франческетти – серьезное врожденное расстройство черепно-лицевого развития, характеризующееся многочисленными аномалиями, которые ограничены головой и лицом. Гипоплазия костей лица, особенно челюстной и скуловой системы, является общей чертой.
Эта болезнь почти полностью отсекает ребенка от общества, делая его жизнь еще более тяжелой и болезненной.
Синдром Франческетти – это генетическое заболевание, вызванное мутацией генов, помогающих правильному развитию костей и лицевых тканей. Этот белок важен для развития лица как в утробе матери, так и после рождения. Мутация приводит к уменьшению его количества, изменяет клетки формирующие черты лица.
Нарушения в развитии костной ткани, вызывают сильную асимметрию, из-за чего слуховой и зрительный аппарат приобретают постоянные структурные патологии.
Распространение
Редкое генетическое заболевание, затрагивает приблизительно 1 из 50 000 человек.
Причины
Развитие заболевания связано с нарушениями внутриутробного развития на 6-7 неделе.
Расстройство возникает из-за мутаций гена TCOF1 или POLR1C, POLR1D. Это аутосомно-доминантное состояние, что означает, что одна копия измененного гена в каждой клетке достаточна, чтобы вызвать расстройство.
Около 60 процентов случаев являются результатом новых мутаций и встречаются у людей, не имеющих истории семейного расстройства. Чаще, человек наследует измененный ген от пострадавшего родителя.
Когда синдром Франческетти вызывается мутациями гена POLR1C, состояние имеет аутосомно-рецессивный характер наследования. Такое наследование означает, что обе копии гена в каждой клетке имеют мутации.
Родители индивидуума с аутосомно-рецессивным состоянием имеют одну копию мутированного гена, но обычно не проявляют признаков и симптомов состояния.

Мутации гена TCOF1 являются наиболее распространенной причиной расстройства, составляя от 81 до 93 процентов всех случаев.
POLR1C и POLR1D вызывают дополнительные 2 процента случаев.
Белки, полученные из генов TCOF1, POLR1C и POLR1D, играют важную роль в раннем развитии костей и других тканей лица. Они участвуют в получении молекулы, называемой рибосомальной РНК (рРНК). Рибосомальная РНК помогает собрать белковые строительные блоки (аминокислоты) в новые белки, которые необходимы для нормального функционирования и выживания клеток.
Мутации TCOF1, POLR1C, POLR1D уменьшают продукцию рРНК. Исследователи предполагают, что уменьшение количества рРНК может спровоцировать саморазрушение (апоптоз) определенных клеток, участвующих в развитии костей и тканей лица.
Аномальная гибель клеток приводит к специфическим проблемам развития лица, обнаруженным при синдроме Франческетти.

Признаки
Признаки этого расстройства сильно различаются: от почти незаметного до тяжелого. Симптомы синдрома Франческетти часто обнаруживаются на УЗИ во время беременности. Чаще проявляется деформированное и недоразвитое лицо, подбородок.
Нижние веки поражены, скулы, челюсть, уши могут отсутствовать или иметь дефекты. У детей возникают проблемы с дыханием и слухом.

Симптомы таковы:
Челюсти и зубы
- Расщепление неба (неправильная крыша рта);
- Неполное открытие рта;
- Зубные промежутки;
- Маленькая челюсть;
- Углы рта наклонены вниз.
Лицо
- Отсутствует кость бровей;
- Отсутствующие или маленькие скулы;
- Нисходящие наклонные глаза;
- Нижнее веко опускающееся;
- Расширение области рта.
Уши
- Характеризуется отсутствующими, маленькими или необычно сформированными ушами;
- Отсутствие слухового прохода;
- Уши расположены слишком близко к области шеи.
Потеря слуха вызвана дефектами трех небольших костей в среднем ухе, которые передают звук, или из-за недостаточного развития слухового прохода.

Общие симптомы
- Проблемы с глотанием;
- Нарушения зрения. Часто имеют глаза, которые наклонены вниз, редкие ресницы, выемка на нижних веках, называемая coloboma;
- Потеря слуха;
- Затрудненное дыхание;
- Проблемы с речью;
- Деформации рук.
Некоторые рождаются с отверстием в крыше рта, называемым волчьей пастью. В тяжелых случаях недоразвитие костей лица ограничивает дыхательные пути пострадавшего ребенка, вызывая потенциально опасные для жизни проблемы с дыханием.
Люди с расстройством обычно имеют нормальный интеллект.

Рекомендации по лечению и коррекции
В настоящее время нет препаратов для лечения синдрома Франческетти. Лечение направлено на конкретные потребности каждого ребенка или взрослого. Необходим многодисциплинарный медицинский подход. Главная проблема – нарушение проходимости дыхательных путей, глотание, зрение, слух.
Новорожденным требуется немедленная трахеостомия, если воздушные пути деформированы. Процедура предполагает размещение трубки непосредственно в области шеи, горла.
Потеря слуха рассматривается как усиление костной проводимости путем размещения имплантов, рекомендуется речевая терапия, образовательные программы.
Во многих случаях необходима черепно-лицевая реконструкция. Хирургия выполняется для восстановления расщепления неба, восстановления челюсти или других костей черепа. Конкретные хирургические процедуры и возраст при проведении операции зависят от тяжести аномалий, общего состояния здоровья, личных предпочтений.
На фотографии девушка до и после пластической хирургии. Поэтому не отчаивайтесь и не опускайте руки. Всегда возможно найти способ улучшить свой внешний вид.
Костные трансплантаты устанавливаются, чтобы помочь создать скулы. Некоторые из операций откладываются до достижения ребенком 5 лет и старше.
В некоторых случаях невозможно полностью исправить деформации лица, поэтому врачи слегка уменьшают проявление симптомов улучшая качество жизни пациента.
Попытки восстановить слух хирургическим путем (для коррекции структуры слуховых косточек) не показали положительного результата, поэтому рекомендуется использовать слуховые аппараты, выбранные для индивидуальных патологий внутреннего и среднего уха.
Новые методы
Есть несколько возможных методов лечения, которые исследуются. Ученые ищут способы ингибировать протеин p53, который помогает организму убивать нежелательные клетки. У людей с синдромом Франческетти, p53 аномально активируется, что приводит к потере определенных клеток, вызывает симптоматику.
Есть предположение, что ингибирование продуцирования р53 (блокирование его активации) может помочь лечить пораженных людей. Необходимы дополнительные исследования, чтобы определить, эффективен ли этот тип лечения и безопасен ли он.
Исследователи также изучают использование стволовых клеток, обнаруженных в жировой ткани, которые используются при операциях у людей с черепно-лицевыми патологиями.
Ранние исследования показали, что хирургические результаты улучшаются с использованием этих стволовых клеток, помогают стимулировать восстановление роста пораженных участков. Однако подобная терапия все еще экспериментальная и противоречивая.

Прогноз и продолжительность жизни
Дети, рожденные с тяжелыми дефектами, которые влияют на дыхание, умирают вскоре после рождения. Некоторые могут быть спасены при немедленной трахеостомии для возможности дыхания.
Врачи, вскоре после рождения, часто выполняют операцию, которая двигает челюсть в правильное положение, чтобы ребенок смог дышать сам по себе раньше. Это улучшает ожидаемую продолжительность жизни для новорожденных.
Обычно люди с синдромом Франческетти становятся работоспособными взрослыми с нормальным интеллектом. В некоторых случаях прогноз зависит от конкретных симптомов и тяжести заболевания. Очень тяжелые случаи могут вызывать перинатальную смерть из-за патологии дыхательных путей.
Основной проблемой синдрома является социальная адаптация.
Ожидаемая продолжительность жизни для людей с синдромом Франческетти такая же, как и у всех, если все осложнения будут устранены на ранней стадии (дыхательные пути, решение проблем кормления, слуха, зрения). Они имеют нормальный интеллект и могут вести продуктивную жизнь.
По Д-р J Jaranson
Понравилась статья? Поделись с друзьями:
Источник