
Генетический анализ синдром прадера вилли

Генетическое исследование, позволяющее выявить причинный фактор – мутацию (отсутствие или недостаточное функционирование некоторых генов (или их частей) на 15-й отцовской хромосоме (синдром Прадера – Вилли) или мутацию материнского генетического материала аналогичного генетического региона (синдром Ангельмана, а также мутация (выпадение одного из участков) 22-й хромосомы, ведущая к гипоплазии вилочковой и паращитовидной желез (синдром Ди Джорджи).
Синонимы русские
Тест на синдромы “смежных генов”, FISH-тест на генетические аномалии, FISH-анализ на Прадера – Вилли, Ангельмана, Ди Джорджи.
Синонимы английские
The test for syndromes of “adjacent genes”, FISH analysis on Prader-Willy syndrome (PWS), FISH analysis for Angelman syndrome (AS), FISH analysis for Di George syndrome (DGS), FISH-test for genetic abnormalities.
Метод исследования
Дифференциальное окрашивание хромосом.
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
- Исследование проводится в состоянии сытости, не рекомендуется сдавать кровь на данное исследование натощак.
- Исключить (по согласованию с врачом) прием антибактериальных и химиотерапевтических препаратов в течение 14 дней до исследования.
- Исследование рекомендуется проводить не ранее чем через 2 недели после перенесенных инфекционных/острых воспалительных заболеваний.
Общая информация об исследовании
Цитогенетический анализ проводится методом флуоресцентной гибридизации in situ (FISH, от англ. fluorescence in-situ hybridization), который является одним из самых современных методов диагностики хромосомных аномалий. Подробнее с методом можно ознакомиться здесь (https://helix.ru/kb/item/12-052).
Одним из преимуществ FISH-теста является его способность определять микроделеции, которые не выявляются с помощью классического кариотипирования или полимеразной цепной реакцией (ПЦР). Это имеет особое значение при диагностике микроделеционных синдромов, например Прадера – Вилли, Ангельмана, Ди Джорджи. Делеции – это хромосомные перестройки, при которых происходит потеря участка хромосомы. Микроделеции часто характеризуются сложным клиническим и поведенческим фенотипом, возникающим в результате дисбаланса нормального количества генов, расположенных в этом конкретном сегменте хромосом.
Микроделеционные синдромы определяются как группа клинически узнаваемых расстройств, характеризующихся небольшой утратой хромосомного сегмента, охватывающего несколько соседних генов, каждый из которых потенциально может вносить свой вклад в фенотип. Эти синдромы называют еще синдромами “смежных генов”. Большинство клинически значимых микроделеций, по-видимому, происходят спорадически, но в ряде случаев может обнаруживаться наследственная связь.
Велокардиофациальный синдром – наиболее частая известная интерстициальная делеция, обнаруженная у человека с частотой 1 из 4000 живорождений. Большинство потерь являются результатом события de novo, и только около 5-10% наследуются аутосомно-доминантным способом. Для диагностики этого синдрома используется несколько диагностических меток, включая синдром (аномалию) Ди Джорджи (DGS).
Синдром Ди Джорджи связан с делецией 22q11.2 и встречается у 1:3000-1:20000 новорождённых, с одинаковой частотой как у мальчиков, так и у девочек. Клинически он проявляется нарушениями иммунной системы, вплоть до тяжелого иммунодефицита из-за недоразвитости или отсутствия тимуса, гипопаратиреозом, пороками сердца, расщелиной неба, умственной отсталостью разной степени выраженности, катарактой (помутнение хрусталика). Также могут быть пороки развития мочевыделительной системы, проблемы с питанием. Больные, как правило, низкого роста и с характерными чертами лица (подбородок и рот маленькие, переносица расширена).
Синдром Прадера – Вилли (75% случаев) и 80% случаев возникновения синдрома Ангельмана связаны с микроделецией 15q11-13. Идентифицированы различные молекулярные механизмы, приводящие к этой потере. Примечательно, что при этом каждый из синдромов может сопровождаться и другими мелкими “молекулярными поломками”.
Синдром Прадера-Вилли (PWS) представляет собой сложное мультисистемное расстройство, характеризующееся множеством клинических проявлений. Встречается патология у 1:12000-1:15000 новорождённых, которые в большинстве случаев рождаются преждевременно. Как правило, пациенты с данной аномалией небольшого роста, с короткими конечностями, имеют типичные черты лица: небольшой лоб, миндалевидной формы глаза, недоразвитую верхнюю челюсть, тонкую верхнюю губу и опущенные углы рта. С самого рождения наблюдается гиперфагия (неконтролируемое потребление пищи) и в результате – ожирение, тяжелая мышечная гипотония. Также характерны гипогонадизм (синдром, сопровождающийся недостаточностью функций половых желез и нарушением синтеза половых гормонов) с недостаточным ростом во время полового созревания, нарушения терморегуляции и дневная сонливость.
Синдром Прадера – Вилли рассматривается как многоэтапное расстройство, характеризующееся тремя различными фазами:
- Первая, “гипотоническая фаза”, характеризуется разной степенью выраженности гипотонии во время неонатального периода и раннего младенчества, слабым криком, гипотермией, гипогенитализмом и слабым рефлексом сосания. В течение первого года дети с PWS дружелюбные, спокойные и ласковые.
- “Гиперфагическая фаза” обычно начинается в 1-2 года, характеризуется повышенным аппетитом, ранним началом детского ожирения, физической бездеятельностью, сниженной болевой чувствительностью, нарушенной терморегуляцией, психомоторной отсталостью, трудностью сочетанной речи и умственными нарушениями. Одновременно с изменением структуры питания меняется и поведение детей – оно становится социально неадекватным и неправильно эмоционально окрашенным (например, немотивированные вспышки гнева), появляются упрямство, лабильность настроения, импульсивность, беспокойство и обсессивно-компульсивные симптомы.
- Третья фаза – “подростковый возраст и взрослая жизнь”. На этом этапе преобладают проблемы со здоровьем, связанные с ожирением (например, сколиоз, гипертония, гиперхолестеринемия, остеопороз и т.д.). Около 10% подростков и взрослых имеют серьезные психиатрические проблемы. Примечательно, что психотические эпизоды у пациентов с PWS имеют много общих черт, включая острое начало, полиморфную и колеблющуюся симптоматику с тревогами, агитацией, аномальными верованиями и слуховыми галлюцинациями. Эти эпизоды классифицируются как острый циклоидный психоз.
Синдром Ангельмана (AS) встречается примерно у 1:10000-30000 новорождённых, и с первых лет жизни наблюдается задержка умственного и неврологического развития. Способность к невербальному общению сохранена, больные понимают больше, чем способны выразить.
К типичным чертам при данной патологии относятся брахицефалия (короткоголовость), микроцефалия (недоразвитость черепа, маленькая голова), большой рот с широко расставленными зубами, нижнечелюстной прогнатизм (выпячивание нижней челюсти, затрагивающее более низкую треть лица), глубокие и голубые глаза и гипопигментация, практически постоянная улыбка. “Типичное лицо” становится очевидным в возрасте от одного года до четырех лет, и с увеличением возраста происходит огрубление и усиление характерных черт. У пациентов с AS наблюдается гипотония с гипертонией конечностей и высокий риск развития сколиоза. Все пациенты имеют умственную отсталость с различными речевыми расстройствами. С первых лет жизни отмечаются различные неврологические нарушения (например, дрожание уголков рта, выталкивание языка, немотивированное хлопанье руками, тики). У 80% детей в возрасте от 1 мес. до 5 лет встречаются эпилептические припадки. Их трудно контролировать, и они могут быть разнообразными, начиная от атипичного отсутствия судорог, тонико-клонических припадков, миоклонических приступов и тонических приступов до эпилептического статуса. Модели ЭЭГ, наблюдаемые у пациентов с синдромом Ангельмана, очень характерны и наблюдаются у пациентов с/без судорог и могут играть важную диагностическую роль в соответствующем клиническом контексте. Уже через 10 недель после рождения могут наблюдаться приступы легко спровоцированного продолжительного смеха. Дети, как правило, гиперактивны, имеют пониженную способность концентрировать внимание и различные нарушения сна. С возрастом походка становится медленная, атаксическая и жесткая с характерным положением поднятых рук с изгибами запястий и локтей (дерганная походка марионетки). Характерны нарушения координации, хаотичные движения рук и ног. Люди с AS плохо переносят душные помещения и повышенную температуру. Кроме классических проявлений синдрома, существуют разнообразные вариации, связанные с различными генетическими причинами. Например, более мягкая эпилепсия отмечена у пациентов со спонтанной мутацией материнской копии, которая вызывает отсутствие преобразования в мозге копии гена UBE3A.
FISH-анализ позволяет достоверно диагностировать общеизвестные микроделеционные синдромы и является стандартным диагностическим методом при данной патологии. Исследование позволяет установить этиологию заболевания и сделать прогноз по дальнейшему его течению.
Для чего используется исследование?
- Для диагностики микроделеций.
Когда назначается исследование?
- Пренатальная диагностика при подозрении на наличие отклонений в развитии плода.
- Постнатальная диагностика генетической патологии у ребенка при наличии соответствующих клинических признаков.
- При планировании последующих беременностей, если в семье есть ребенок с микроделеционным синдромом.
Что означают результаты?
Положительный результат говорит о наличии в исследуемом образце микроделеции.
Отрицательный результат указывает на отсутствие искомой микроделеции, но не может достоверно опровергать заболевание, так как синдром может быть вызван другим генетическим сбоем (например, однородительской дисомией, транслокацией родительских хромосом). В процентном соотношении вероятность этого значительно меньше, но все же возможна.
Синдром Прадера-Вилли:
- встречается у 1:25000- 1:10000 новорождённых;
- с помощью обычного исследования хромосомного состава кариотипа выявить данную патологию невозможно.
Синдром Ангельмана:
- встречается примерно у 1:10000 новорождённых;
- с первых лет жизни наблюдается задержка умственного и неврологического развития.
Синдром Ди Джорджи:
- встречается у 1:3000-1:20000 новорождённых, в равной доле как у мальчиков, так и у девочек;
- характерна триада симптомов: гипоплазия тимуса и паращитовидной железы, врождённый порок сердца;
- 5-10% – аутосомно-доминантный тип наследования.
Важные замечания
FISH-тест часто используется совместно с другими методами молекулярной и цитогенетической диагностики.
Также рекомендуется
[40-006] Беременность – Пренатальный скрининг трисомий I триместра беременности (синдром Дауна)
[16-001] Исследование кариотипа
[40-007] Беременность – Пренатальный скрининг трисомий II триместра беременности
Кто назначает исследование?
Педиатр, врач-генетик, эндокринолог, невролог.
Литература
- Shaffer, L.G., Ledbetter, D.H., and Lupski, J.R. 2001, Molecular cytogenetics of contiguous gene syndromes: mechanisms and consequences of gene dosage imbalance. In: The Metabolic & Molecular Bases of Inherited Disease, C. R. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle (Eds.), McGraw-Hill, Medical Publishing Division, New York, St. Louis, San Francisco, Auckland, Bogota, Caracas, Lisbon, London, Madrid, Mexico City, Milan, Montreal, New Delhi, San Juan, Singapore, Sudney, Tokyo, Toronto, 1291.
- Microdeletions and Molecular Genetics, Annick VOGELS and Jean-Pierre FRYNS. Center for Human, University of Leuven, Herestraat 49, B-3000 Leuven, Belgium, 2004-02.
- Di George, A. 1965, A new concept of the cellular basis of immunity. J. Pediatr., 67, 907.
- Clinical spectrum and molecular diagnosis of Angelman and Prader-Willi syndrome patients with an imprinting mutation.Saitoh S, Buiting K, Cassidy SB, Conroy JM, Driscoll DJ, Gabriel JM, Gillessen-Kaesbach G, Glenn CC, Greenswag LR, Horsthemke B, Kondo I, Kuwajima K, Niikawa N, Rogan PK, Schwartz S, Seip J, Williams CA, Nicholls RD, Am J Med Genet. 1997;68(2):195.
Источник
Что такое синдром Прадера — Вилли?
Синдром Прадера — Вилли (СПВ) — это генетическое заболевание, которое встречается примерно у одного из каждых 15 000 новорожденных.
Синдром Прадера — Вилли поражает мужчин и женщин с одинаковой частотой и затрагивает все расы и этнические группы. СПВ признана наиболее распространенной генетической причиной опасного для жизни детского ожирения.
Данный синдром был впервые описан швейцарскими врачами Андреа Прадером, Алексисом Лабхартом и Генрихом Вилли в 1956 году на основании клинических характеристик девяти обследованных детей.
Общие характеристики, определенные в первоначальном отчете, включали маленькие руки и ноги, аномальный рост и состав тела (маленький рост, очень низкая мышечная масса тела и раннее детское ожирение), мышечная гипотония (низкий мышечный тонус (слабость мышц)) при рождении, ненасытный голод, сильное ожирение и умственная отсталость.
Это видео даст краткое понимание проявлений болезни:
Синдром Прадера — Вилли является результатом аномалии хромосомы 15, и постановка точного диагноза сегодня основывается на генетических анализах.
Каковы симптомы синдрома Прадера — Вилли?
Симптомы синдрома Прадера — Вилли, вероятно, связаны с дисфункцией части мозга, называемой гипоталамус. Гипоталамус — это небольшой эндокринный орган у основания мозга, который играет важную роль во многих функциях организма, включая регулирование голода и сытости, температуры тела, боли, баланса сна и бодрствования, водного баланса, эмоций и фертильности.
Хотя считается, что дисфункция гипоталамуса приводит к появлению симптомов синдрома Прадера — Вилли, еще не ясно, как генетическая аномалия вызывает дисфункцию гипоталамуса.
У людей с данным синдромом симптомы со временем меняются. В целом, есть два основных этапа симптомов, связанных с СПВ:
Ранний период жизни
Младенцы с СПВ являются гипотоническими или «гибкими», с очень низким мышечным тонусом. Для них типичен слабый крик и плохой сосательный рефлекс. Детей с синдромом Прадера — Вилли обычно не могут кормить грудью и они часто нуждаются в искусственном вскармливании.
У детей могут быть проблемы с ростом и развитием, если трудности с кормлением не подвергаются тщательному мониторингу и лечению. По мере взросления этих детей сила и мышечный тонус в целом улучшаются. Моторные функции развиты, но, как правило, выполняются с задержкой.
Малыши, как правило, вступают в период, когда они могут легко начать набирать вес до того, как проявят повышенный интерес к еде.
Детство и взросление
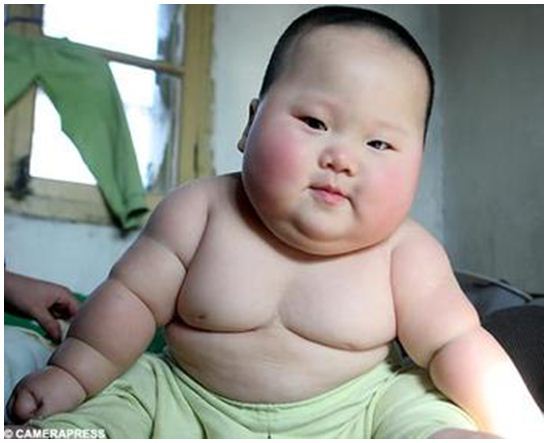
Нерегулируемый аппетит и легкое увеличение веса (см. фото) характеризуют более поздние стадии синдрома Прадера — Вилли.
Эти признаки чаще всего начинаются в возрасте от 3 до 8 лет, но различаются по началу и интенсивности.
Люди с синдромом Прадера — Виллии испытывают недостаток в нормальных подсказках голода и сытости.
Как правило, они не могут контролировать потребление пищи и переедают, если их не контролировать.
Поведение в голоде очень распространено. Кроме того, скорость метаболизма у людей с данным синдромом ниже, чем обычно. Без лечения эта комбинация проблем приводит к патологическому ожирению и его многочисленным осложнениям.
Помимо ожирения, у ребенка с синдромом Прадера — Вилли может быть множество других симптомов. Дети с IQ от низкого нормального до умеренной умственной отсталости обычно имеют когнитивные проблемы. Те, у кого нормальный IQ, обычно имеют проблемы с обучаемостью.
Другие проблемы и признаки могут включать:
- дефицит гормона роста/низкий рост;
- маленькие руки и ноги;
- сколиоз;
- нарушения сна с чрезмерной дневной сонливостью;
- высокий болевой порог;
- апраксия/диспраксия речи и бесплодие.
Поведенческие трудности могут включать в себя обсессивно-компульсивные симптомы, проблемы с кожей и трудности с контролем эмоций. Взрослые с синдромом Прадера— Вилли подвергаются повышенному риску психических заболеваний. СПВ является расстройством широкого спектра, и симптомы варьируют по степени тяжести и частоты встречаемости среди людей.
Хромосомы и гены: основы
Чтобы понять генетику СПВ, необходимо иметь общее представление о хромосомах и генах.
Хромосомы представляют собой крошечные структуры, которые присутствуют почти в каждой клетке нашего тела. Это пакеты генов, которые мы наследуем от наших родителей. Гены содержат все подробные инструкции, необходимые нашему телу для роста, развития и функционирования — нашу ДНК.
Определенные гены направляют наши клетки на производство белков, ферментов и других необходимых веществ. Каждый из наших многочисленных генов находится на определенной хромосоме. Большинство клеток нашего тела содержат 46 хромосом — 23 унаследованы от нашей матери и 23 от нашего отца. (Яйца и сперматозоиды обычно содержат всего 23 хромосомы, потому что это те клетки объединяются в зачатии и обеспечивают ребенку нужное количество хромосом.)
22 пары хромосом помечены числом, основанным на их размере (хромосома 1 — самая большая пара, а хромосома 22 — почти наименьшая), и 2 хромосомы в каждой пронумерованной паре содержат одинаковые гены (один набор у матери и одну от отца). Изменения, которые вызывают синдром Прадера — Вилли, происходят в паре, известной как хромосома 15. 23-я пара хромосом обозначена как пара половых хромосом. Эта пара определяет пол ребенка: XX для девочки, XY для мальчика.
Изменения или ошибки в генах и хромосомах распространены при образовании яйцеклеток и сперматозоидов. Некоторые из этих генетических изменений не будут иметь эффекта, когда ребенок зачат; некоторые вызовут выкидыш; а некоторые спровоцируют, например, синдром Прадера — Вилли, вызвав значительные различия в том, как ребенок развивается и функционирует.
В то время как многие генетические нарушения вызваны изменением одного гена и могут передаваться от родителя к ребенку, СПВ куда более сложен.
Что вызывает синдром Прадера — Вилли (причины)?
Синдром Прадера — Вилли возникает в результате отсутствия активного генетического материала в определенной области хромосомы 15 (15q11-q13). Обычно люди наследуют одну копию хромосомы 15 от своей матери и одну от своего отца. Гены вызывающие СПВ активны только в хромосоме, полученной от отца.
При синдроме Прадера — Вилли генетический дефект, вызывающий неактивность хромосомы 15 от отца (отцовская хромосома 15), может возникнуть по трем следующим причинам:
- СПВ по исключению: чаще всего часть хромосомы 15, которая была унаследована от отца, отсутствует или удалена в этой критической области. Эта небольшая делеция встречается примерно в 70% случаев и обычно не выявляется при обычном генетическом анализе, таком как амниоцентез.
- СПВ по однородительской дисомии: еще около 30% случаев происходят, когда человек наследует две хромосомы 15 от своей матери и ни один от своего отца. Это тип наследование называется однородительской дисомией (UPD).
- СПВ по импрессии мутации: наконец, в очень небольшом проценте случаев (1-3%) небольшая генетическая мутация при синдроме Прадера — Вилли приводит к тому, что генетический материал отцовской хромосомы 15 хоть и присутствует, но неактивен.
Как эти генетические дефекты вызывают симптомы, наблюдаемые при синдроме Прадера — Вилли?
Хромосомы 15 одни из самых сложных областей генома человека. Хотя были достигнуты значительные успехи в понимании и характеристике генетических изменений, связанных с синдромом Прадера — Вилли, точный механизм, с помощью которого отсутствие функционального генетического материала приводит к симптомам, связанным с СПВ, не понятны.
Ученые активно изучают нормальную роль генетических последовательностей в области СПВ и то, как их потеря влияет на гипоталамус и другие системы организма.
Существуют ли различия в степени тяжести СПВ в зависимости от генетического подтипа?
Могут быть некоторые тонкие различия в признаках СПВ на основе генетического подтипа: например, лица с делециями могут быть светлокожими со светлыми волосами по сравнению с другими членами семьи и быть более восприимчивыми к припадкам и судорогам; а те, у кого есть проблемы с СПВ по однородительской дисомии, могут быть подвержены более высокому риску психических заболеваний в молодом подростковом возрасте.
В целом, однако, существует значительное совпадение между различными генетическими подтипами. Вероятно, что тысячи генов за пределами области СПВ, которые демонстрируют нормальные различия между индивидуумами, также вносят значительный вклад в вариабельность симптомов синдрома Прадера — Вилли между теми, у кого есть расстройство.
Как диагностируется СПВ?
Данный синдром диагностируется с помощью анализа крови, который выявляет генетические аномалии, специфичные для синдрома Прадера — Вилли — так называемый «анализ метилирования ДНК». Метод FISH (флуоресцентная гибридизация in-situ) идентифицирует синдром путем делеции, но не диагностирует другие формы СПВ.
Анализ на метилирование ДНК идентифицирует все типы этого синдрома и является предпочтительным методом исследования для диагностики. Если анализ на метилирование проводится первым, может потребоваться дополнительное исследование, чтобы определить, вызван ли СПВ отцовской делецией, UPD или импринтинговой мутацией.
В случаях, когда существует подозрение на наличие импринтинговой мутации, кровь также может быть взята у родителей.
Каковы риски синдрома Прадера — Вилли в будущем?
СПВ по исключению и UPD являются случайными явлениями и, как правило, не связаны с повышенным риском рецидива в будущих беременностях. В случае импринтинг-мутации синдром Прадера — Вилли может в семье повториться. Семьи с опасениями по поводу их риска СПВ должны поговорить с генетическим консультантом.
Есть ли лекарство от синдрома Прадера — Вилли?
В настоящее время нет никакого лечения от синдрома Прадера — Вилли, и большинство исследований на сегодняшний день было направлено на лечение определенных симптомов данного синдрома.
Для многих людей, затронутых этим расстройством, устранение некоторых из самых сложных аспектов синдрома, таких как ненасытный аппетит и ожирение, будет представлять собой значительное улучшение качества жизни и способности жить независимо.
Источник