
Генетика 3 с синдромом дауна

Генетика синдрома Дауна: кариотипКлинический диагноз синдрома Дауна обычно не представляет никаких трудностей. Тем не менее для подтверждения диагноза и предоставления базы для генетического консультирования необходимо кариотипирование. Хотя различия в конкретных вариантах кариотипа, ответственных за синдром Дауна, обычно имеют небольшое влияние на фенотип пациента, они существенны для определения риска повторения. Трисомия 21 при синдроме Дауна. Примерно у 95% всех пациентов с синдромом Дауна выявляют трисомию хромосомы 21, вызванную мейотическим нерасхождением 21 пары хромосом, как обсуждалось в предыдущей главе. Уже отмечено, что риск иметь ребенка с трисомией 21 увеличивается с возрастом матери, особенно после 30 лет. Мейотическая ошибка, ответственная за трисомию, обычно происходит в ходе материнского мейоза (около 90% случаев), преимущественно в первом делении, но около 10% случаев происходит в отцовском мейозе, обычно во втором делении. Робертсоновская транслокация при синдроме Дауна. Около 4% пациентов с синдромом Дауна имеют 46 хромосом, одна из которых — робертсоновская транслокация между хромосомой 21q и длинным плечом одной из других акроцентрических хромосом (обычно хромосомы 14 или 22). Транслоцированная хромосома заменяет одну из нормальных акроцентрических хромосом, и кариотип пациента с робертсоновской транслокацией между хромосомами 14 и 21 – 46,XX/XY,rob(14;21)(ql0;ql0),+21. Такая хромосома может также быть определена как der(14;21), на практике используют обе номенклатуры. В действительности пациенты с робертсоновской транслокацией, включающей хромосому 21, трисомны по генам, расположенным в длинном плече 21q. В отличие от стандартной трисомии 21, транслокационный синдром Дауна не показывает никакой связи с возрастом матери, но имеет сравнительно высокий риск повторения в семьях, если один из родителей, особенно мать, — носитель транслокации. По этой причине для точного генетического консультирования важно кариотипирование родителей и, возможно, других родственников.
Носители робертсоновской транслокации, включающей хромосомы 14 и 21, имеют только 45 хромосом; одна 14 и одна 21 отсутствуют и заменены транслоцированной хромосомой. Теоретически возможны шесть типов гамет, но три из них не могут привести к жизнеспособному потомству. Три типа гамет жизнеспособные, нормальные, сбалансированные и несбалансированные, имеющие как транслоцированную, так и нормальную хромосому 21. В комбинации с нормальной гаметой это может приводить к зачатию ребенка с транслокационным синдромом Дауна. Теоретически эти три типа гамет производятся в равных количествах, таким образом, теоретический риск ребенка с синдромом Дауна должен быть 1 к 3. Тем не менее расширенные популяционные исследования показали, что несбалансированные хромосомные наборы появляются только у 10-15% потомства матерей и только у нескольких процентов потомства отцов, несущих транслокации, включающие хромосому 21. Транслокация 21q21q при синдроме Дауна. Хромосомная транслокация 21q21q — хромосома, сформированная из двух длинных плеч хромосомы 21; бывает у нескольких процентов пациентов с синдромом Дауна. Считают, что они появляются как изохромосомы, а не робертсоновские транслокации. Большинство таких случаев возникают постзиготически, соответственно, риск повторения низкий. Тем не менее особенно важно убедиться, не является ли родитель носителем (возможно, мозаичным) данной транслокации, поскольку все гаметы носителя такой хромосомы должны также содержать 21q21q хромосому, с двойной дозой генетического материала хромосомы 21, или не иметь хромосомы 21 совсем. Потенциальное потомство, следовательно, неизбежно имеет или синдром Дауна, или нежизнеспособную моносомию 21. Мозаичные носители имеют повышенный риск повторения, таким образом, пренатальная диагностика необходима при всех последующих беременностях. Мозаичный синдром Дауна. Около 2% пациентов с синдромом Дауна — мозаики, обычно с популяциями нормальных клеток и с трисомией 21. Фенотип может быть мягче, чем при типичной трисомии 21. Вообще существует широкая изменчивость в фенотипах мозаичных пациентов, вероятно, отражая различные пропорции трисомных клеток у эмбриона на ранних стадиях развития. Возможно, пациенты с установленным мозаичным синдромом Дауна отражают только клинически более серьезные случаи, поскольку в легких случаях кариотипирование менее вероятно. Частичная трисомия 21 при синдроме Дауна. Очень редко синдром Дауна диагностируют у пациентов, имеющих трисомию только по части длинного плеча хромосомы 21, и еще реже выявляют пациентов с синдромом Дауна без цитогенетически видимой хромосомной аномалии. Такие случаи представляют определенный интерес, поскольку могут указывать, какая область хромосомы 21, вероятно, ответственна за специфические компоненты фенотипа синдрома Дауна и какие области могут утраиваться, не вызывая фенотипических проявлений. Хотя хромосома 21 содержит только несколько сотен генов, попытки согласовывать тройную дозу специфических генов со специфическими аспектами фенотипа синдрома Дауна пока имеют ограниченный успех. Наиболее примечательной стала идентификация области, критической для пороков сердца, наблюдаемых примерно у 40% пациентов с синдромом Дауна. Поиск конкретных генов, существенных для проявления фенотипа синдрома Дауна, среди случайно находящихся рядом с ними в хромосоме 21, — главная задача современных исследований, особенно на мышах в качестве модели. Потенциально перспективное направление — исследование генно-инженерных мышей с дополнительной дозой генов из хромосомы 21 человека (или даже с полной копией хромосомы 21). Такие мыши могут проявлять фенотипические аномалии в поведении, функциях мозга и формировании сердца. – Также рекомендуем “Причины синдрома Дауна. Риск рождения ребенка с трисомией 21″ Оглавление темы “Хромосомные аномалии”:
|
Источник
Высокий риск синдрома Дауна?

Синдром Дауна не является болезнью, это патология которую невозможно предотвратить и вылечить. У плода с синдромом Дауна в 21-й паре хромосом имеется третья дополнительная хромосома, в итоге их количество составляет не 46, а 47. Синдром Дауна наблюдается у одного из 600-1000 новорожденных от женщин в возрасте после 35. Причина, по которой это происходит, до конца не выяснена. Врач из Англии Джон Лэнгдон Даун первым описал этот синдром в 1866 году, а в 1959 году французский профессор Лежен доказал, что это связано с генетическими изменениями.
Известно, что половину хромосом дети получают от матери, а половину – от отца. Поскольку нет ни одного эффективного метода лечения синдрома Дауна, болезнь считается неизлечимой, можно принять меры и при желании родить ребенка здоровым, обратиться в медико-генетическую консультацию, где на основании хромосомного анализа родителей будет определено, родится ребенок здоровым или с синдромом Дауна.
В последнее время такие дети рождаются чаще, связывают это с поздним замужеством, с планированием беременности в возрасте 40 лет. Также считается, что если бабушка родила свою дочь после 35, то внуки могут родиться с синдромом Дауна. Хотя дородовая диагностика – сложный процесс обследования, её проведение очень необходимо для того, чтобы была возможность прервать беременность.
Что же являет собой синдром Дауна. Обычно он может сопровождаться задержкой моторного развития. Такие дети имеют врожденные пороки сердца, патологию развития органов желудочно-кишечного тракта. 8% больных с синдромом Дауна болеют лейкемией. Медикаментозное лечение может стимулировать психическую деятельность, нормализовать гормональный дисбаланс. С помощью физиотерапевтических процедур, массажа, лечебной гимнастики можно помочь ребенку приобрести навыки необходимые для самообслуживания. Синдром Дауна связан с генетическим нарушением, но не всегда это приводит к нарушению физического и умственного развития ребенка. Такие дети, а в будущем взрослые люди могут участвовать во всех сферах жизни, некоторые из них становятся актерами, спортсменами и могут заниматься общественными делами. Как будет развиваться человек с данным диагнозом зависит во многом от того окружения в котором он растет. Хорошие условия, любовь и забота способствуют полноценному развитию.
Таблица риска синдрома Дауна, по возрастам
Вероятность синдрома Дауна зависит от возраста матери, но его можно выявить генетическим тестом на ранних стадиях беременности, а в некоторых случаях ультразвуком. Вероятность наличия у ребенка синдрома Дауна при рождении ниже, чем на более ранних стадиях беременности, т.к. некоторые плоды с синдромом Дауна не выживают.
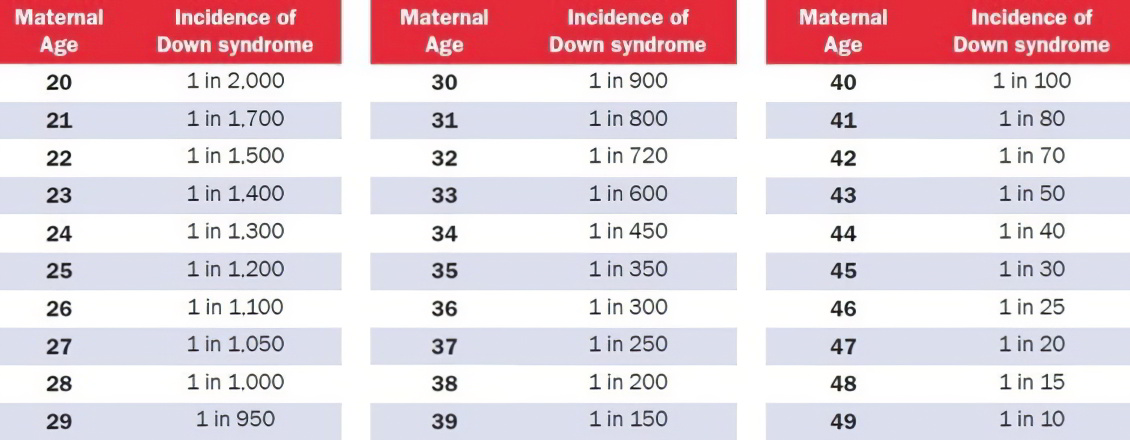
Какой риск считается низким, а какой – высоким?
В Израиле риск синдрома Дауна считается высоким, если он выше, чем 1:380 (0.26%). Всем, кто находится в этой группе риска нужно пройти проверку околоплодных вод. Этот риск приравнивается к риску у тех женщин, которые забеременели в возрасте 35 лет и старше.
Риск ниже, чем 1:380 считается низким.
Но надо учитывать, что эти границы могут быть плавающими! Так, например, в Англии, высоким уровнем риском считается риск выше 1:200 (0.5%). Это происходит по той причине, что одни женщины считают риск 1 к 1000 – высоким, а другие 1 к 100 – низким, так как при таком риске у них шанс на рождение здорового ребенка равен 99%.
Факторы риска синдрома Дауна, Эдвардса, Патау
Основными факторами риска являются возраст (особо значимо для синдрома Дауна), а также воздействие радиации, некоторых тяжелых металлов. Следует учитывать, что даже без факторов риска плод может иметь патологию.
Как видно из графика, зависимость величины риска от возраста наиболее значима для синдрома Дауна, и менее значима для двух других трисомий:
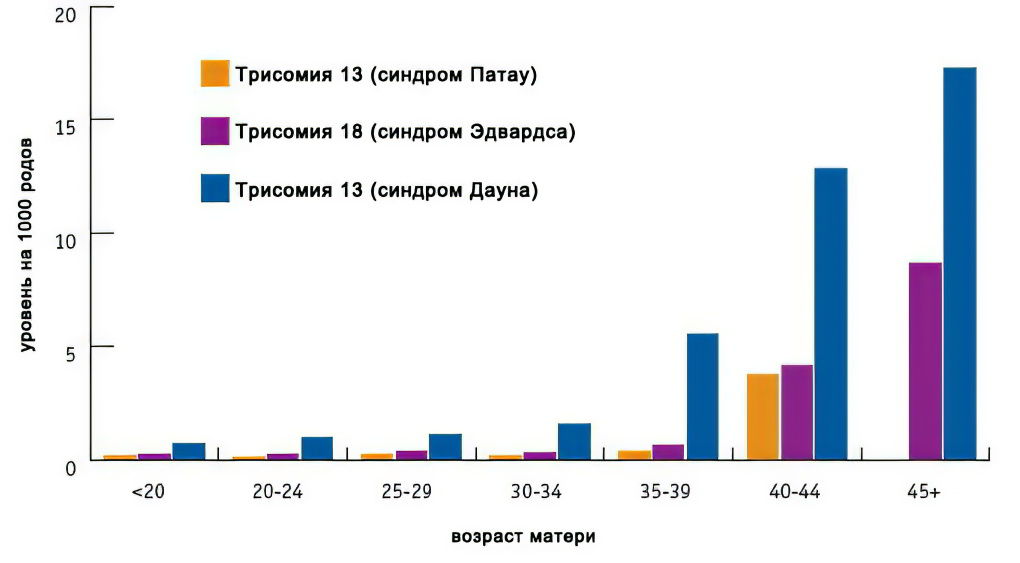
Скрининг риска синдрома Дауна
На сегодняшний день всем беременным, кроме полагающихся анализов рекомендуется проходить скрининговый тест для выявления степени риска синдрома Дауна по рождению ребенка и врожденным порокам плода. Наиболее продуктивное обследование бывает на 11 неделе + 1 день или на 13 неделе + 6 дней при копчико-теменном размере эмбриона от 45 мм до 84 мм. Беременная женщина может пройти обследование, и использовать для этого специфическое УЗИ.
Более точный диагноз ставится при помощи биопсии ворсин хориона и исследования амниотической жидкости, которая забирается с помощью специальной иглы непосредственно из плодного пузыря. Но каждая женщина должна знать, что такие методы сопряжены с риском осложнений беременности таких как выкидыш, инфицирования плода, развития тугоухости у ребенка и многое другое.
Полный комбинированный скрининг I – II триместра беременности позволяет выявить врожденные пороки у плода. Что же включает данный тест? Во-первых, необходимо ультразвуковое исследование в 10-13 недель беременности. Расчет риска производится по определению наличия носовой кости, по ширине шейной складки плода, где скапливается подкожная жидкость в первом триместре беременности.
Во вторах берется анализ крови на хорионический гонадотропин в 10-13 недель и на Альфа-фетопротеин в 16-18 недель. Данные комбинированного скрининга обрабатываются по специальной компьютерной программе. Учеными предложена новая методика скрининга – объединение оценки результатов, полученных в ходе исследований в первом и во втором триместрах. Это позволяет обеспечить единую оценку риска возникновения синдрома Дауна при беременности.
Для первого триместра используются результаты определения РАРР-А и измерения толщины воротникового пространства, а для второго триместра – используются сочетания АФП, неконъюгированного эстриола, ХГ и ингибина-А. Применение интегральной оценки для скринингового обследования позволяет после инвазивных вмешательств снижать частоту прерывания беременности для плодов с нормальным кариотипом по результатам цитогенетической диагностики.
Интегральное и биохимическое тестирование для скрининга синдрома Дауна позволяет дополнительно выявлять больше случаев хромосомных аномалий. Это способствует предотвращению нежелательных прерываний беременности, возникающих в результате амниоцентеза или биопсии ворсин хорион.
Автор статьи: Мочалов Павел Александрович | д. м. н. терапевт
Образование:
Московский медицинский институт им. И. М. Сеченова, специальность – “Лечебное дело” в 1991 году, в 1993 году “Профессиональные болезни”, в 1996 году “Терапия”.
Наши авторы
Источник
1. Синдром Дауна Рассмотрим некоторые хромосомные болезни. Синдром Дауна, трисомия по 21-й хромосоме – самая частая и наиболее хорошо изученная хромосомная болезнь. Частота рождения детей с синдромом Дауна составляет примерно 1:750 и не имеет какой-либо временной, этнической или географической разницы и родителей одинакового возраста. С возрастом (в большей степени матери и в меньшей мере отца) вероятность рождения ребенка с данной патологией существенно возрастает, и в возрасте 45 лет составляет около 3 %. Цитогенетические варианты синдрома Дауна разнообразны. Основную долю составляют случаи полной трисомии 21 как следствие нерасхождения хромосом в мейозе. Наряду с этим известны случаи регулярной трисомии, связанной с транслокацией 21-й хромосомы на другую – 21, 22, 13, 14 или 15-ю хромосому. Почти 50 % транслокационных форм наследуется от родителей носителей и 50 % – вновь возникшие мутации. Соотношение мальчиков и девочек среди новорожденных с синдромом Дауна составляет 1:1. Клиническая картина синдрома Дауна разнообразна: врожденные пороки развития, нарушения постнатального развития нервной системы, иммунодефициты и другие отклонения. Многие симптомы заметны уже при рождении ребенка и дальнейшем проявляются еще более отчетливо. Из черепно-лицевых дизморфий отмечается монголоидный разрез глаз, круглое уплощенное лицо, плоская спинка носа, крупный язык, брахицефалия, деформированные ушные раковины. Так же характерны мышечная гипотония и разболтанность суставов. Часто диагностируются врожденный порок сердца, клинодактилия. Встречаются изменения дерматоглифики в виде четырехпальцевой, или “обезьяньей”, складки на ладони, две кожные складки вместо трех на мизинце. Характерен низкий рост (на 20 см ниже среднего). Диагноз синдрома Дауна ставится на основании клинически на основании сочетания ряда симптомов. Наиболее важные из которых: уплощение профиля лица (90 %), отсутствие сосательного рефлекса (85 %), избыток кожи на шее (80 %), монголоидный разрез глаз (80 %), мышечная гипотония (80 %), разболтанность суставов (80 %), диспластический таз (70 %), деформированные ушные раковины (40 %), клинодактилия мизинца (60 %), четырехпальцевая сгибательная складка (поперечная линия) на ладони (40 %). Большое значение для диагностики имеет задержка умственного и физического развития ребенка. Задержка умственного развития может достигать степени имбицильности, а коэффициент умственного развития у разных детей широко варьируется (IQ от 25 до 75). Больные с синдромом Дауна часто болеют пневмониями, тяжело переносят детские инфекции. У них отмечается недостаток массы тела. Врожденные пороки внутренних органов и недостаточность иммунной системы часто приводят к летальному исходу в первые 5 лет жизни. Дифференциальная диагностика проводится с другими формами хромосомных аномалий и врожденным гипотиреозом. Цитогенетическое исследование показано и при подозрении на синдром Дауна и при клинически установленном диагнозе. В последнем случае это необходимо для прогноза здоровья будущих детей у родителей ребенка и их родственников. Лечебная помощь детям с синдромом Дауна многопланова и неспецифична. Врожденные пороки сердца устраняют оперативно. Постоянно проводится общеукрепляющая терапия, защита от действия вредных факторов внешней среды. Многие больные с трисомией 21 способны вести самостоятельную жизнь, овладевают несложными профессиями, создают семью.
2. Синдром Патау Синдром Патау– трисомия по 13-й хромосоме, выделен в самостоятельную нозологическую форму в 1960 г. в результате генетического исследования у детей с врожденными пороками развития. Обнаружены простые и транслокационные формы трисомии 13, однако клинически и патологоанатомически они неразличимы. Частота синдрома Патау среди новорожденных составляет 1:6000. Соотношение полов при данной патологии близко 1:1. Частое осложнение при вынашивании плода с синдромом Патау – многоводие (50 %). Для заболевания характерны множественные, тяжелые пороки развития головного мозга, мозговой и лицевой частей черепа, внутренних органов. Окружность черепа обычно уменьшена, лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположены и деформированы (80 %). Типичный признак – расщелина верхней губы и неба (70 %). Всегда обнаруживаются пороки внутренних органов в разных комбинациях: пороки сердца (80 %), незавершенный поворот кишечника (40 %), кисты почек (42 %), аномалии внутренних половых органов (73 %), дефекты поджелудочной железы (43 %). Часто наблюдается полидактилия кистей (50 %) и их флексорное положение(44 %). Дети с синдромом Патау практически всегда имеют глубокую идиотию. Клиническая диагностика основывается на сочетании характерных пороков развития. Однако решающий фактор в диагностике – исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших больных, с целью составления прогноза для будущих детей в семье. Лечебные мероприятия неспецифичны: общеукрепляющее лечение, тщательный уход, профилактика простудных и инфекционных болезней. В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы жизни, но некоторые больные живут до нескольких лет.
3. Синдром Клайнфельтера Синдром Клайнфельтераотносится к группе полисомий по половым хромосомам. Заболевание включает в себя случаи полисомии, при которых имеется не менее двух Х-хромосом и не менее одной Y-хромосомы. Наиболее часто (примерно 1:600) встречается синдром Клайнфельтера с набором 47,XXY.Этот синдром является и наиболее типичным клинически. Варианты полисомии с большим числом Х – и Y-хромосом (XXXY,XYY,XXXXY,XXYY) встречаются редко. Присутствие Y-хромосомы определяет формирование мужского пола. До периода полового созревания мальчики развиваются почти нормально. Вызываемый добавочной Х-хромосомой генетический дисбаланс проявляется клинически в период полового созревания в виде недоразвития семенников и вторичных мужских половых признаков. Мужчины с синдромом Клайнфельтера обычно имеют высокий рост, астеническое или евнухоидное телосложение, слабое оволосение лица, подмышечных впадин и лобка. Выявляется умственная отсталость легкой и средней степени, а в четверти случаев гинекомастия. Больные бесплодны (азооспермия, олигоспермия).
Источник