
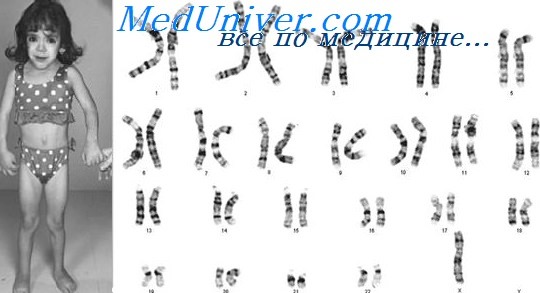
Генотип с синдромом шерешевского тернера

Синдром Тернера: причины, диагностика, лечениеЭтиология и встречаемость синдрома Тернера. Синдром Тернера — панэтническое заболевание, вызываемое полным или частичным отсутствием второй Х-хромосомы у женщин. Его встречаемость составляет от 1 на 2000 до 1 на 5000 среди живорожденных девочек. Около 50% случаев синдрома Тернера вызваны кариотипом 45,Х, 25% — структурной аномалией второй Х-хромосомы, и 25% — мозаицизмом 45,Х. Моногамия по Х-хромосоме может возникать при нарушении перехода половой хромосомы в одну из гамет или при потере половой хромосомы в зиготе или раннем эмбрионе. Наиболее частая причина кариотипа 45,Х — нарушение передачи отцовской половой хромосомы в гамету; 70-80% пациенток с кариотипом 45,Х оплодотворены сперматозоидом с недостающей половой хромосомой. Вероятная причина мозаицизма 45,Х — утрата половой хромосомы в клетке раннего эмбриона. Патогенез синдрома ТернераМеханизм формирования клинической картины синдрома Тернера у девочек с моносомей по Х-хромосоме мало понятен. Х-хромосома содержит множество локусов, не подвергающихся инактивации, некоторые из них необходимы для функционирования яичников и женской фертильности. Хотя для развития овоцита достаточно одной Х-хромосомы, для их функционирования необходимы две Х-хромосомы. Следовательно, при отсутствии второй Х-хромосомы, овоциты у плодов и новорожденных с синдромом Тернера деградируют, а их яичники атрофируются в полоски соединительной ткани. Генетическая основа других признаков синдрома Тернера, например, кистозной гигромы, лимфоотека, широкой грудной клетки, пороков сердца, аномалий почек и нейросенсорной тугоухости, не определена, но, возможно, отражает гаплонедостаточность одного или более Х-сцепленных генов, в норме не подвергающихся инактивации у женщин. Фенотип и развитие синдрома ТернераХотя плоды с кариотипом 45,Х встречаются при 1-2% всех беременностей, заканчиваются живорожденным младенцем с кариотипом 45,Х менее 1% родов. Учитывая мягкий фенотип, наблюдаемый у пациенток с синдромом Тернера, обращает на себя внимание столь высокий показатель выкидышей, указывая на важность второй половой хромосомы для внутриутробного выживания. Все пациенты с синдромом Тернера имеют низкий рост, и более 90% —дисгенезию яичников. Дисгенезия яичников достаточно выраженная, поэтому только у 10-20% пациентов пубертат развивается самостоятельно (молочные железы и лобковое оволосение) и только 2-5% имеют самостоятельные менструации. Большинство больных также имеет физические аномалии, например, складчатую шею, низкий рост волос на затылке, широкую грудную клетку, пороки сердца и почек, нейросенсорную тугоухость, отеки кистей и стоп и диспластичные ногти. Почти у 50% пациенток отмечают двустворчатый аортальный клапан, и, следовательно, повышенный риск расширения и расслаивающей аневризмы корня аорты; почти у 60% имеются аномалии почек и повышенный риск дисфункции почек.
У большинства пациенток интеллектуальное развитие нормальное. Если есть интеллектуальная задержка, обычно обнаруживается структурная аномалия Х-хромосомы. Больные с синдромом Тернера обычно стеснительны и замкнуты (см. главу 6). Помимо осложнений, вызванных врожденными аномалиями, у женщин с синдромом Тернера чаще встречаются остеопоротические переломы костей, тиреоидит, сахарный диабет I и II типа, колиты и заболевания сердечно-сосудистой системы. Причины сахарного диабета, нарушений щитовидной железы и колитов неясны. Вероятно, в основном за остеопороз и повышенную встречаемость атеросклероза, ишемических заболеваний сердца и инсульты ответственна эстрогенная недостаточность, хотя возможно, что сахарный диабет усиливает влияние недостатка эстрогенов на сердечно-сосудистую систему. Особенности фенотипических проявлений синдрома Тернера: Лечение синдрома ТернераЕсли рост пациентки с синдромом Тернера отстает ниже 5-го процентиля, обычно назначают соматотропный гормон, применяемый до достижения костного возраста, соответствующего 15 годам. В среднем это лечение дает прибавку роста в 10 см; прибавка роста тем меньше, чем позже начато лечение гормоном роста. Параллельная терапия эстрогенами уменьшает эффективность гормона роста. Терапию эстрогенами обычно начинают в возрасте 14-15 лет, для обеспечения развития вторичных половых признаков и снижения риска остеопороза. Прогестерон для вызывания менструаций добавляют к лечению либо во время первого вагинального кровотечения, либо на втором году терапии эстрогенами. Кроме того, медицинская помощь обычно включает следующие виды диагностики: эхокардиографию с целью оценки расширения корня аорты и клапанных заболеваний сердца, УЗИ почек для исключения врожденных аномалий и нагрузочный тест с глюкозой для выявления сахарного диабета. У пациенток с полной дисгенезией яичников не бывает спонтанных овуляций и беременности. Однако если позволяет состояние сердечно-сосудистой системы и почек, женщины с синдромом Тернера могут иметь детей в случае подсадки оплодотворенной яйцеклетки. Риски наследования синдрома ТернераСиндром Тернера не связан с возрастом матери или отца. Хотя описано несколько случаев повторения в семье, обычно синдром Тернера спорадический, и эмпирический риск повторения при будущих беременностях не превышает общепопуляционного. Если синдром Тернера заподозрен на основе УЗИ плода, например, выявлена кистозная гигрома, диагноз следует подтвердить кариотипированием ворсин хориона или амниоцитов. Среди спонтанно менструирующих женщин с синдром Тернера описано несколько случаев наступления беременности. У одного из трех родившихся детей отмечены врожденные аномалии, такие как врожденные пороки сердца, синдром Дауна и spina bifida. Очевидно, повышенный риск врожденных аномалий может быть следствием выборочного смещения в публикациях, так как беременность редко встречается при синдроме Тернера. Если повышенный риск аномалий — реальный факт, то его причина неизвестна. Пример синдрома Тернера. Л.В., девушка 14 лет, направлена в клинику эндокринологии для обследования по поводу отсутствия вторичных половых признаков (менструаций и развития молочных желез). Хотя Л.В. родилась с небольшой массой тела, она была полностью здорова и имела нормальный интеллект. Другие члены семьи аналогичных проблем не имели. Данные обследования оказались нормальными, за исключением низкого роста, I стадии полового развития по Таннеру и широкой грудной клетки с широко расположенными сосками. После краткого обсуждения возможных причин задержки роста и полового развития врач назначил определение уровня фолликулостимулирующего гормона, гормона роста (соматотропина), анализ костного возраста и анализ хромосом. Эти тесты показали нормальный уровень гормона роста, повышение уровня фолликулостимулирующего гормона и аномальный кариотип (45,Х). Врач дал заключение, что у девочки синдром Тернера. Ей был назначен гормон роста; годом позже она начала терапию эстрогенами и прогестероном для стимуляции развития вторичных половых признаков. – Также рекомендуем “Пигментная ксеродерма: причины, диагностика, лечение” Оглавление темы “Генетика”:
|
Источник
… четкой связи возникновения синдрома Шерешевского-Тернера с возрастом и какими-либо заболеваниями родителей не выявлено.
Синдром Шерешевского-Тернера – это хромосомная болезнь, проявляющаяся сочетанием сердечно-сосудистых, эндокринных, репродуктивных, психосоциальных нарушений и пороков развития; частота синдрома составляет около 1 : 2500 новорожденных девочек. Хромосомные аномалии при данном синдроме проявляются в виде отсутствия одной из двух Х-хромосом, делеции части одной Х-хромосомы или транслокации в пределах одной Х-хромосомы, возможны также различные мозаичные варианты, когда хромосомный набор частично сохранен. У некоторых пациенток возможно полное или частичное наличие Y-хромосомы, что является фактором риска в отношении развития гонадобластомы.
К основным клиническим характеристикам синдром Шерешевского-Тернера относятся:
• задержка роста;
• первичная овариальная недостаточность;
• ряд врожденных аномалий и метаболических нарушений.
Клинические и патофизиологические особенности синдрома Шерешевского-Тернера (СШТ):
нарушения роста и формирования скелета: низкий рост (наблюдается в 95–100 % случаев СШТ, средняя величина конечного роста варьирует от 140 до 147 см), короткая шея, cubitus valgus (девиация локтевых суставов), короткие метакарпальные кости и укорочение IV и V пальцев кистей рук, деформация Медалунга, сколиоз, genu valgum (искривления костей голени), характерное лицо с микрогнатией, высокое готическое небо (специфическое строение неба нередко обусловливает носовой или высокий оттенок голоса); иногда встречаются аномалии роста зубов, которые требуют лечения у ортодонта; у пациенток с СШТ выше частота встречаемости врожденной дисплазии тазобедренного сустава, а у 10–20 % девочек развивается сколиоз;
нарушения герминативного эпителия: первичная недостаточность гонад, гонадобластома, (первичная овариальная недостаточность наблюдается в 95–98 % случаев СШТ; дефицит эстрогенов способствует развитию остеопороза у женщин с СШТ, что вызывает высокую частоту переломов в характерных местах – запястье, позвоночник, шейка бедра);
лимфостаз: с возрастом чаще всего лимфатические отеки исчезают, однако могут периодически появляться при нагрузках; с лимфостазом у пациенток с СШТ связывают наличие таких особенностей фенотипа, как крыловидные складки шеи, ногтевая дисплазия (в т.ч. ее тяжелая форма), низкий рост волос на шее, аномалии строения ушных раковин (ротированные ушные раковины), отечность кистей или стоп;
врожденные пороки: нарушения сердечно-сосудистой системы (коарктация аорты – 11 %, бикуспидальный аортальный клапан – 16 %; основная причина смерти больных с СШТ – сердечно-сосудистая патология, в частности разрыв дилатированной аорты), почечные и реноваскулярные дефекты (пороки чашечно-лоханочной системы – 20%, подковообразная почка, при которой две почки срастаются, обычно нижними полюсами – 10 %, мальротация и другие аномалии расположения – 5%; др. варианты пороков развития включают одно- или двухстороннюю гипоплазию почек, удвоение лоханки и мочеточников, изменение числа почечных вен или артерий, а также их нетипичное расположение), множественные пигментные невусы, косоглазие, птоз, эпиканта, антимонголоидный разрез глаз; патология органа слуха у пациенток с СШТ характеризуется высокой частотой средних отитов, манифестация которых приходится на возраст от 1 до 6 лет, в результате средних отитов часто возникает кондуктивная тугоухость; для взрослых женщин с СШТ более характерна прогрессирующая нейросенсорная тугоухость, снижение слуха наблюдается в основном после 35 лет;
метаболические и физиологические нарушения: тиреоидит Хашимото, гипотиреоз, алопеция, витилиго, гастроинтестинальные нарушения, в т.ч. целиакия (4–6 %), нарушение толерантности к углеводам (является риском развития сахарного диабета 2-го типа), эссенциальная артериальная гипертензия; у пациенток с СШТ отмечена большая частота ожирения, чем в популяции.
Диагноз «синдром Шерешевского-Тернера» устанавливается на основании исследования кариотипа; показаниями к кариотипированию являются сочетания особенностей фенотипа и наличия пороков различных органов с задержкой роста в детском возрасте, с задержкой пубертата у подростков, с первичной или вторичной аменореей на фоне повышения гонадотропных гормонов у взрослых женщин.
Показаниями для изучения кариотипа плода могут служить обнаруженные у него при УЗИ следующие признаки:
• утолщение затылочного изгиба;
• водянка мозга;
• коарктация аорты;
• аномалии строения левых отделов сердца;
• брахицефалия;
• аномалии строения почек;
• внутриутробная задержка развития плода;
• сопутствующее нарушение в гормональном статусе у матери.
Наблюдение пациенток с СШТ должно осуществляться при совместном сотрудничестве таких специалистов, как генетики, кардиологи, педиатры, терапевты, эндокринологи, гинекологи, отоларингологи и др., и проводиться в течение всей жизни. Постоянный мониторинг пациенток с СШТ осуществляется по следующей схеме:
• девочки до 5 лет: оценка социальных умений в возрасте 4–5 лет;
• девочки школьного возраста: ежегодная оценка функции печени и щитовидной железы (св.Т4, ТТГ), познавательного и социального развития; каждые 2–5 лет – скрининг на целиакию (антитела к трансглутаминазе); по необходимости обследование у стоматолога и ортодонта (если возраст > 7 лет);
• девочки старшего возраста и взрослые: оценка пубертатного развития и психосексуальной адаптации, определение уровней липидов и глюкозы в сыворотке крови натощак; ежегодно оценивают функцию печени и щитовидной железы; по показаниям проводят скрининг на целиакию;
• пациентки всех возрастов: ежегодно проводят кардиологическое обследование (при впервые выявленном СШТ – осмотр кардиолога для исключения врожденных пороков сердца, четкая визуализация сердца, аортального клапана, дуги аорты и легочных вен: провдят МРТ [младенцам и маленьким детям не проводится] и ЭхоКГ, ЭКГ, измерение артериального давления; в дальнейшем – обследование каждые 5 – 10 лет, при выявленной патологии сердечно-сосудистой системы схему наблюдения и лечение определяет кардиолог); каждые 4–5 лет – аудиологическое исследование.
Необходимо помнить, что у девочек от 0 до 4 лет должно осуществляться обследование (скриниг) для исключения дисплазии тазобедренных суставов; если девочке > 1 года – необходима оценка зрения у детского офтальмолога; у пациенток всех возрастов также долженн осуществляться скрининг на выявление сколиоза или кифоза, остеопороза (с 18 лет осуществляют контроль минеральной плотности костей). Следует отметить, что наиболее частыми причинами врачебного наблюдения у взрослых женщин с СШТ являются артериальная гипертензия, нарушения углеводного и липидного обменов, ожирение.
Одной из основных задач лечения больных с СШТ в детском и подростковом возрасте являются увеличение конечного роста и формирование вторичных половых признаков с последующим установлением регулярного менструального цикла. Лечение низкорослости при СШТ на сегодняшний день включает применение рекомбинантного гормона роста: ежедневное п/к введение в супрафизиологической дозе 0,05 мг/кг/сут перед сном; чем продолжительнее ростстимулирующее лечение в препубертатный период, тем больше конечный рост; терапию гормоном роста прекращают, когда костный возраст пациентки становится равен 15 годам, а скорость роста падает до 2 см в год. Заместительная терапия половыми стероидами при СШТ используется для максимально точного имитирования полового развития здоровой девочки. При заместительной терапии половыми стероидами и проведении ростстимулирующей необходим мониторинг у педиатра-эндокринолога каждые 6 месяцев.
Источник
Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Тернера (синдром Шерешевского-Тёрнера, синдром Бонневи-Ульриха, Синдром 45, Х0) является следствием полного или частичного отсутствия одной из двух половых хромосом, фенотипически определяется женский пол. Диагноз основывается на клинических проявлениях и подтверждается исследованием кариотипа. Лечение зависит от проявлений и может включать оперативное лечение при пороках сердца, и часто терапию гормоном роста при низкорослости и заместительную терапию эстрогеном при отсутствии пубертата.
Клинически впервые описан отечественным эндокринологом Н. А. Шерешевским в 1926 г. Цитогенетически синдром верифицирован в 1959 г. Популяционная частота 1 : 5000 лиц женского пола.
[1], [2], [3], [4], [5], [6]
Код по МКБ-10
Q96 Синдром Тернера
Q96.8 Другие варианты синдрома Тернера
Q96.9 Синдром Тернера неуточненный
Эпидемиология синдрома Шерешевского-Тёрнера
Синдром Шерешевского-Тёрнера встречается с частотой примерно 1 /4000 живорождений и является наиболее распространенной аномалией половых хромосом у женщин. В то же время 99 % беременностей с кариотипом плода 45, X заканчиваются спонтанным абортом.
[7], [8], [9], [10], [11]
Что вызывает синдром Шерешевского-Тёрнера?
Примерно у 50 % пациенток с синдромом Шерешевского-Тёрнера отмечается кариотип 45, X; примерно в 80 % случаев теряется отцовская Х-хромосома. Большая часть оставшихся 50 % мозаична (например, 45, Х/46, XX или 45, Х/47, XXX). Среди мозаичных пациентов фенотип может варьировать от типичного для синдрома Тернера до нормального. Иногда у девочек с синдромом Шерешевского-Тёрнера могут отмечаться одна нормальная Х-хромосома и одна Х-кольцевая хромосома; для того чтобы произошло формирование кольцевой хромосомы, она должна потерять по участку с короткого и длинного плеча. Некоторые девочки с синдромом Тернера могут иметь одну нормальную Ххромосому и одну изохромосому, состоящую из двух длинных плеч Х-хромосомы, которая сформировалась после потери короткого плеча. У таких девочек отмечается склонность иметь многие из фенотипических черт синдрома Тернера; таким образом кажется, что деления короткого плеча Х-хромосомы играет важную роль в формировании фенотипа синдрома Шерешевского-Тёрнера.
Патогенез синдрома Шерешевского-Тёрнера
Отсутствие или изменение половой хромосомы приводит к нарушению созревания яичников, отсутствию или позднему частичному половому созреванию и бесплодию.
Симптомы синдрома Шерешевского-Тёрнера
У многих новорожденных отмечаются только очень легкие проявления; однако у некоторых наблюдаются выраженная дорсальная лимфедема кистей рук и ступней, а также лимфедема или кожные складки на задней поверхности шеи. Другие распространенные аномалии включают крыловидные складки шеи, широкую грудную клетку и втянутые соски. У пораженных девочек отмечается низкий рост по сравнению с членами семьи. Менее частыми признаками являются низкая линия роста волос на задней поверхности шеи, птоз, множественные пигментные невусы, короткие четвертые пястная и плюсневая кости, выступающие подушечки пальцев с завитками на концах пальцев, а также гипоплазия ногтей. Также отмечаются cubitus valgus (вальгусное отклонение в локтевом суставе).
Распространенные аномалии сердца включают коарктацию аорты и двухстворчатый клапан аорты. Часто с возрастом развивается гипертензия, даже в отсутствие коарктации. Нередко встречаются аномалии почек и гемангиомы. Иногда в ЖКТ обнаруживают телеангиэктазию, с развивающимися желудочнокишечными кровотечениями или потерей белка.
Дисгенезия гонад (яичники замещаются двухсторонними тяжами фиброзной стромы с отсутствием развивающихся половых клеток) отмечается у 90 % больных, приводя к отсутствию пубертата, отсутствию увеличения грудных желез, аменорее. В то же время у 5-10 % пораженных девочек спонтанно происходит менархе, и очень редко пораженные женщины фертильны и имеют детей.
Задержка умственного развития отмечается редко, но у многих пациентов наблюдается снижение некоторых перцептивных возможностей и вследствие этого низкие оценки в невербальных тестах и по математике, даже несмотря на то, что баллы, полученные за вербальный компонент тестов на интеллект, средние или даже высокие.
- Задержка роста, часто с рождения (100%).
- Гонадальный дисгенез с аменореей и стерильностью.
- Лимфатический отёк тыла кистей и стоп (40%).
- Широкая грудная клетка с комбинированной деформацией грудины.
- Широко расставленные, гипоплазированные и инвертированные соски (80%).
- Аномальные по форме и оттопыренные ушные раковины (80%).
- Низкий уровень роста волос.
- Короткая шея с избытком кожи и крыловидными складками (80%).
- Cubitus valgus(70%).
- Узкие, гипервогнутые и вдавленные ногти (70%).
- Врождённые пороки почек (60%).
- Тугоухость (50%).
- Врождённые пороки сердца и аорты (коарктация аорты и патология клапанов, расширение и расслоение аорты) (20-40%).
- Идиопатическая артериальная гипертензия (АГ) (27%).
Диагностика синдрома Шерешевского-Тёрнера
У новорожденных диагноз можно заподозрить при наличии лимфедемы или крыловидной складки шеи. В отсутствие этих изменений диагноз у некоторых детей выявляют позже на основании низкого роста, аменореи и отсутствия пубертата. Диагноз подтверждается исследованием кариотипа. Для обнаружения врожденных пороков сердца показано проведение эхокардиографии или МРТ.
Цитогенетический анализ и исследования с Y-специфичным зондом проводят всем лицам с дисгенезией гонад для исключения мозаицизма с наличием клеточной линии с кариотипом 46, XY (45, X/46XY). У таких пациентов обычно женский фенотип с различными чертами синдрома Тернера. Они находятся в группе повышенного риска по развитию злокачественных новообразований гонад, особенно гонадобластомы, поэтому для профилактики сразу после определения диагноза следует удалить гонады.
[12], [13], [14], [15], [16], [17]
Физическое обследование
Диагноз ставят на основании характерной клинической картины: короткая шея с избытком кожи и крыловидными складками у новорождённых девочек, лимфатический отёк кистей и/или стоп, врождённые пороки левого сердца или аорты (особенно коарктация аорты), задержка роста и полового развития в пубертатный период у девочек.
Лабораторные исследования
Цитогенетическое исследование верифицирует отсутствие Х-хромосомы или её структурные изменения.
Какие анализы необходимы?
Лечение синдрома Шерешевского-Тёрнера
Не существует специфического лечения генетического нарушения. Коарктацию аорты обычно лечат оперативно. При других пороках сердца показаны динамическое наблюдение и хирургическая коррекция при необходимости. При лимфедеме применяют компрессионные чулки.
Терапию рекомбинантным человеческим гормоном роста могут начинать, если рост менее 5 перцентилей, начальная доза составляет 0,05 мг/кг подкожно один раз в день. Как правило, в возрасте 12-13 лет для инициации пубертата необходима заместительная терапия эстрогеном в виде конъюгированных эстрогенов по 0,3 мг внутрь или микронизированного эстрадиола по 0,5 мг один раз в день. После этого назначают оральные контрацептивы, содержащие прогестин, для поддержания вторичных половых признаков. Гормон роста продолжают вводить вместе с терапией половыми гормонами до закрытия зон роста, когда терапию гормоном роста прекращают. Продолжение заместительной терапии половыми гормонами помогает достичь оптимальную плотность костей и развитие скелета.
Синдром Шерешевского-Тёрнера лечится с помощью коррекции низкорослости путем проведения курсов человеческого соматропина, а также формирования вторичных половых признаков путем назначения эстрогенов.
Какой прогноз имеет синдром Шерешевского-Тёрнера?
Синдром Шерешевского-Тёрнера имеет благоприятный для жизни и интеллектуального развития прогноз. Неблагоприятный для деторождения.
Источник