
Глазные щели узкие слуховой проход отсутствует это синдром

Трисомии, синдром (болезнь) Дауна, Патау, Эдвардса у ребенка, детей
Наиболее часто у человека встречаются трисомии по 21-й, 13-й и 18-й паре хромосом.
Синдром (болезнь) Дауна у детей, ребенка
Синдром (болезнь) Дауна — синдром трисомии 21 — самая частая форма хромосомной патологии у человека (1:750). У мальчиков и девочек патология встречается одинаково часто.
Достоверно установлено, что дети с синдромом Дауна чаще рождаются у пожилых родителей. Если возраст матери 35-46 лет, то вероятность рождения больного ребенка возрастает до 4,1 %. Возможность возникновения повторного случая заболевания в семье с трисомией хромосомы 21 составляет 1-2 % (с возрастом матери риск увеличивается).
Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб, широкое, плоское лицо. Типичны эпикант, запавшая спинка носа, косой (монголоидный) разрез глазных щелей, пятна Брушфильда (светлые пятна на радужке), толстые губы, утолщенный язык с глубокими бороздами, выступающий изо рта, маленькие, округлой формы, низко расположенные ушные раковины со свисающим завитком, недоразвитая верхняя челюсть, высокое нёбо, неправильный рост зубов, короткая шея.
Из пороков внутренних органов наиболее типичны пороки сердца (дефекты межжелудочковой или межпредсердной перегородок, фиброэластоз и др.) и органов пищеварения (атрезия двенадцатиперстной кишки, болезнь Гиршпрунга и др.). Среди больных с синдромом Дауна с более высокой частотой, чем в популяции, встречаются случаи лейкемии и гипотиреоза. У маленьких детей резко выражена мышечная гипотония, а у детей старшего возраста часто обнаруживается катаракта. С самого раннего возраста отмечается отставание в умственном развитии. Средняя продолжительность жизни при синдроме Дауна составляет 36 лет.
Синдром Патау у детей, ребенка
Синдром Патау — синдром трисомии 13 — встречается с частотой 1:6000. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае болезни Дауна.
При синдроме Патау наблюдаются тяжелые врожденные пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы. У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезенки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Характерна задержка умственного развития.
Большинство больных детей с синдромом Патау (98 %) умирают в возрасте до года, оставшиеся в живых страдают глубокой идиотией.
Синдром Эдвардса у детей, ребенка
Синдром Эдвардса — синдром трисомии 18 — встречается с частотой примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребенка составляет 0,7 %. У девочек встречается значительно чаще, чем у мальчиков.
Дети с трисомией 18 рождаются с низким весом, хотя сроки беременности нормальные или даже превышают норму. Фенотипические проявления синдрома Эдвардса многообразны. Наиболее часто отмечаются аномалии мозгового и лицевого черепа, мозговой череп долихоцефалической формы. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщен и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и легочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса.
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 мес, до года доживает лишь один ребенок из десяти; оставшиеся в живых — глубокие олигофрены.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 25 мая 2015;
проверки требуют 16 правок.
Синдром Э́двардса (синдром трисомии 18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом (John H. Edwards). Популяционная частота примерно 1:3000 в США, и 1:5000 в мире на 2016 год. Дети с трисомией в 18 хромосоме чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21[3] и 13[4]. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков. Выживание после года жизни составляет около 5–10%[5].
Причины заболевания[править | править код]
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Проявления синдрома[править | править код]
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Прогноз[править | править код]
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления[править | править код]
Частота появления синдрома Эдвардса составляет ~ 1:7000 зачатий и 1:8000 рождений живых детей. Риск рождения больного ребёнка увеличивается с возрастом, особенно, если мать болеет диабетом.
Вариации[править | править код]
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикреплённая к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
См. также[править | править код]
- Анеуплоидия
- Хромосомные болезни
- Синдром Дауна
- Синдром Патау
Примечания[править | править код]
- ↑ база данных Disease ontology (англ.) — 2016.
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ Синдром Дауна
- ↑ Синдром Патау
- ↑ Genetics Home Reference. Trisomy 18 (англ.). Genetics Home Reference. Дата обращения 20 сентября 2019.
Ссылки[править | править код]
- https://rh-conflict.narod.ru/student/lectures/hrombol.htm
Источник
- Главная
- Врожденные пороки
- Синдром Эдвардса
Абсолютное большинство людей знают, что есть такое заболевание, как синдром Дауна. Кто-то знает, что это хромосомное заболевание, а кто-то полагает, что это психическое или неврологическое отклонение от нормы. Но мало кому известно, что хромосомных заболеваний куда больше, и одно из них – синдром Эдвардса. Возможно это обусловлено коротким периодом жизни детей, родившихся с синдромом Эдвардса, и относительно низким процентным отношением таких детей к здоровым детям (1 ребенок с синдромом на 7000 живых новорожденных и на 6000 всех новорожденных).
ИнформацияВысок процент естественного невынашивания беременности с генетически неполноценным плодом и процент прерывания беременностей после установления диагноза плода.
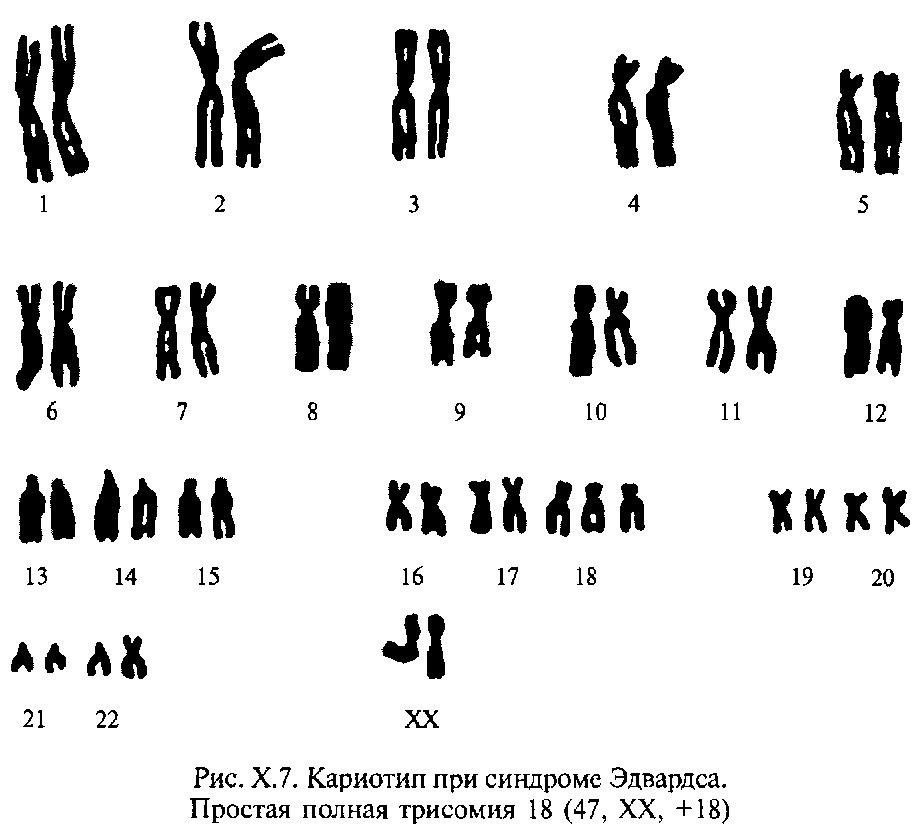
Что это такое?
Синдром Эдвардса – это тяжелое хромосомное заболевание, которое называется также трисомией 18 хромосомы. Заболевание сопровождается тяжелейшими патологиями всех органов и множественными пороками развития. Чаще всего с синдромом Эдвардса рождаются девочки: их в три раза больше, чем мальчиков с той же патологией. Возможно это связано с тем, что природный механизм жизнеобеспечения женских плодов «работает» лучше.
ДополнительноУченые-медики Австралии исследовали плаценты и пришли к выводу, что они имеют разное строение и по-разному осуществляют свои защитные функции в зависимости от пола вынашиваемого плода. Ранее статистика отмечала, что вынашиваемость девочек в пороками развития выше, чем мальчиков с аналогичными отклонениями, а смертность среди новорожденных девочек ниже, чем среди мальчиков при прочих равных условиях.
Причины синдрома Эдвардса
Причина синдрома Эдвардса – в наличии дополнительной 18-ой хромосомы в кариотипе зиготы. В норме для диплоидного набора две хромосомы. В организме человека все клетки диплоидны, то есть содержат в ядре две хромосомы. Гаплоидны – имеют в ядре одну хромосому – только половые клетки (гаметы). Гаплоидность половых клеток обусловлена тем, что при оплодотворении каждая клетка привносит свой набор из 23 хромосом. Таким образом у плода будет необходимые 46 хромосом – по половине от каждого родителя.
Иногда в момент оплодотворения происходит сбой и формируется неправильное количество хромосом. Эта «неправильность» как раз выступает причиной хромосомных заболеваний: синдрома Эдвардса, синдрома Дауна, синдрома Патау. Все эти заболевания вызывают серьезные отклонения в развитии детей, сказываются на их здоровье и существенно снижают качество жизни пациентов. Все эти дети независимо от степени аномалий неспособны к самостоятельной жизни и нуждаются в постоянном уходе.
ИнформацияИногда нарушается число половых хромосом. Это выступает причиной ряда других патологий. Редко половая клетка уже содержит неправильное число хромосом. Такие заболевания приводя к сексуальному инфантилизму, невозможности вести половую жизнь, бесплодию, но в целом не так катастрофично сказываются на общем здоровье пациентов и позволяют вести практически полноценную жизнь.
Любые хромосомные отклонения связывают с возрастом матери на момент первых родов. Считается, что после 45 лет женщина имеет шанс родить ребенка с рассматриваемой патологией, равный 0,7 %. Тем не менее количество молодых матерей в возрасте 20-30 лет, родивших ребенка с синдромом Эдвардса, выше, чем количество матерей того же возраста, у которых родились дети с синдромами Дауна и Патау. Возраст матери не так выражено связан с такой хромосомной патологией, как синдром Эдвардса. Предположительно риск генетических отклонений у детей, зачатых в зрелом возрасте, связывается с возможными генными мутациями в клетках матери в течение жизни под влиянием неблагоприятных факторов окружающей среды. Однако, это только предположение и не объясняет все случаи рождения детей с хромосомными заболеваниями.
Признаки заболевания
Диагностику детей, рожденных с синдромом Эдвардса, проводят в первую очередь по внешнему виду. Новорожденные с любой хромосомной аномалией выглядят специфично. У каждого заболевания есть собственные фенотипические признаки. Младенцы при нормальной и даже превышающей норму беременности рождаются с низким весом – примерно 2200 г. Вероятно это связано с нарушениями кровообращения и трофики тканей младенца, а также с недостаточностью пуповинного питания во всю длительность беременности.
Фенотипические проявления заболевания очень многообразны:
- аномалии черепа и лицевых костей;
- мозговой череп обычно долихоцефалической – длинной и узкой – формы;
- нижняя челюсть недоразвита, ротовое отверстие неправильное и маленькое;
- глазные щели узкие и короткие, ушные раковины неправильной формы с отсутствующими частями и низко расположены;
- иногда отсутствует слуховой проход либо он очень короткий, из-за чего дети рождаются глухими;
- грудина короткая, широкая, межреберные промежутки существенно уменьшены;
- обычно деформирована стопа – большой палец толстый и короткий, пятка резко выступает, свод провисает.
Возможны другие неспецифические проявления – «заячья губа», «волчья пасть», полидактия, олигодактия и другие отклонения. Неспецифичность означает, что эти отклонения встречаются при других генетических отклонениях.
В ходе подробного морфологического обследования при синдроме Эдвардса у новорожденного выявляются пороки развития сердца и крупных сосудов, патологии других внутренних органов. Без вмешательства пища часто не может проходить по пищеварительному тракту, нарушена способность опорожнять кишечник и мочевой пузырь.
ИнформацияУ всех больных синдромом Эдвардса присутствуют гипоплазия мозжечка (из-за этого движения ребенка раскоординированы и неконтролируемы, что сохраняется на весь период жизни), гипоплазия мозолистого тела (взывает судороги, нарушение сенсорных реакций, неспособность к терморегуляции, малую модуляцию крика), нарушение структур олив – частей продолговатого мозга (из-за этого наблюдаются проблемы с самостоятельным дыханием и кровообращением, в именно остановка дыхания — одна из наиболее частых причин смертности детей с синдромом Эдвардса).
У детей с синдромом Эдвардса также выражена мышечная слабость, ненормальная расслабленность всех мышц, отсутствует реакция на раздражители – свет, звук, прикосновение, не развиты рефлексы, иногда даже сосательный и глотательный. В случае доживания детей до годовалого возраста и старше, что происходит крайне редко, почти никогда, отмечается глубокая умственная отсталость вплоть до идиотии и полное отсутствие интеллекта.
Болезнь Эдвардса на УЗИ
Всем беременным женщинам предписывается пройти обследование на предмет наличия хромосомных отклонений у плода. Синдром Эдвардса до 12 недель никак не диагностируется, хотя отклонения есть с момент оплодотворения яйцеклетки, но начиная с 12 недели возможно обнаружить характерные для этого заболевания симптомы. К ним относятся:
- Брадикардия (нарушение у плода сердцебиения в виде снижения частоты сердечных сокращений),
- Омфалоцеле (наличие в брюшной полости грыж),
- Отсутствие визуально определенных косточек носа,
- Отсутствие в пуповине одной артерии (в норме в пуповине должно быть две артерии),
- Наличие кисты сосудистых сплетений (киста сама по себе опасности для плода не несет и самопроизвольно исчезает к 26 неделе беременности, но она часто возникает на фоне различных генетических заболеваний у плода, в том числе синдрома Эдвардса).
Наличие хотя бы одного из указанных отклонений выступает поводом для направления беременной женщины на дополнительное исследование. Принудительного исследования женщины не проводится, на все диагностические процедуры и манипуляции она должна давать добровольное согласие. Врач обязан разъяснить смысл и важность диагностических процедур в данном случае, а также порядок их проведения.
На более поздних сроках беременности на УЗИ могут быть обнаружены расщепление позвоночника плода вследствие дефекта нервной трубки, увеличение жидкости в воротниковом пространстве, укорочение костей, деформация черепа, изменения в структурах мозга. Эти отклонения свойственным многим хромосомным аномалиям.
УЗИ на выявление хромосомных заболеваний у плода называется скрининговым и проводится врачами, прошедшими специальную подготовку в области генетических аномалий развития. Для этого женщина обычно направляется в иное учреждение для обследования, поскольку не в каждой поликлинике есть необходимое оборудование и специалист с соответствующим уровнем подготовки.
ДополнительноПрограмма скрининга беременных в 1 триместре включает в себя также анализ сыворотки крови, поскольку ни один самый точный прибор УЗИ не может с абсолютной точностью показать наличие или отсутствие заболеваний у плода.
Риск рождения ребенка с хромосомным заболеванием рассчитывается по таким показателям:
- количество ассоциированного с беременностью плазменного протеина А (анализ сыворотки)
- количество свободной β-субъединицы хорионического гонадотропина человека (анализ сыворотки),
- морфологические признаки (УЗИ).
Индивидуальный риск рождения ребенка с генетической аномалией развития 1/100 и выше.
Самый точный диагноз может быть поставлен на основе исследования плодного материала.
Для этого проводится инвазивная процедура. Какая именно процедура будет проведена, зависит от срока беременности:
- В 8-12 недель биопсия ворсин хориона (ткань хориона имеет тот же набор хромосом, что и плод, потому с точностью показывает наличие либо отсутствие генетических отклонений,
- В 14-18 недель амниоцентез (забор и анализ околоплодной жидкости),
- В 18-20 недель и позднее кордоцентоз (забор и анализ пуповинной крови).
Амниоцентез и кордоцентез часто проводятся вместе в ходе одной диагностической процедуры. Это позволяет получить более всесторонние и точные результаты.
Важно В случае постановки диагноза хромосомных нарушений в том числе и синдрома Эдвардса у плода и врожденных пороков развития с неблагоприятным прогнозом для жизни и здоровья ребенка по решению перинатального консилиума и добровольного согласия беременной женщины производится прерывание беременности независимо от срока беременности.
Женщина обязательно должна быть проинформирована о диагнозе плода, о течении диагностированного заболевания, особенностях развития и качестве жизни детей, рожденных с подобной аномалией.
После 16-18 недель беременности проводится тройной скрининговый тест на хромосомные отклонения плода. В ходе биохимического анализа крови оцениваются такие показатели:
- α-фетопротеин (альфа-ФП);
- свободная β-субъединица хорионического гормона человека (бета-ХГЧ);
- эстриол свободный.
Виды синдрома
Выделяют вид полной трисомии синдрома Эдвардса, когда во всех клетках организма ребенка есть дополнительная хромосома (это 95 % всех случаев), мозаичную трисомию, когда дополнительная хромосома обнаруживается не во всех клетках (3 % всех случаев), вид частичной трисомии синдрома Эдвардса, когда часть хромосомы присрелдинена к другой хромосоме (2 % всех случаев). Первый и второй варианты трисомии 18 развиваются в одной из половых клеток родителей до зачатия. Третий вариант развивается после оплодотворения.
Во втором и третьем виде развития синдрома Эдвардса выражены чуть слабее, но также не позволяют ребенку вести полноценную жизнь.
Протекание синдрома Эдвардса
Из-за того, что синдром Эдвардса сопряжен с множественными патологиями развития внутренних органов, прогноз для жизни у него крайне неблагоприятный. В первые три месяца после рождения умирают около 60 % детей. До года доживают около 5-10 %, а до подросткового возраста – менее 1 %. Эти дети – глубокие олигофрены. Они не говорят, практически не понимают обращенную речь (часто среди них встречаются глухие), не запоминают ничего, не координируют свои действия, не контролируют позывы к мочеиспусканию и дефекации, не могут самостоятельно есть (по некоторым данным часть подростков освоили самостоятельный прием пищи без помощи столовых приборов), дети с синдромом Эдвардса практически не ходят или ходят с большим трудом и опорой. У них резко снижена чувствительность, в том числе болевая. В большинстве случаев им трудно переменить положение своего тела. Осмысленная деятельность абсолютному большинству из больных синдромом Эдвардса недоступна. Они не отличают людей друг от друга, их внимание почти никогда не бывает сосредоточенным. Они не могут плакать или смеяться, но отмечались случаи, когда дети с этим заболеванием умели улыбаться и даже позитивно реагировали на ласку.
Наиболее частые причины наступления смерти у детей с синдромом Эдвардса – остановка дыхания или остановка работы сердца.
Лечение и последствия синдрома Эдвардса
Исправление и лечение хромосомных нарушений невозможно. Из-за сильнейших патологий развития практически всего организма детей с синдромом Эдвардса оперативное лечение по восстановлению структур и функционирования внутренних органов почти безрезультатно. Вся медицинская помощь сводится к постоянному контролю за жизнедеятельностью ребенка и поддержки его семьи.
Сразу после рождения лечение направляется на коррекцию тех пороков, которые создают самую явную угрозу жизни ребенка: хирургически восстанавливается проход пищи при аномалиях развития пищевода и / или кишечника, налаживается кормление через зонд, поскольку часто отсутствуют или крайне слабо выражены сосательный и глотательный рефлексы. При необходимо проводится противовоспалительная и антибактериальная терапия.
ИнформацияЕсли прогноз для младенца относительно благоприятный, то принимаются меры по хирургической коррекции пороков сердца и сосудов, устраняются грыжи, устраняется полностью или частично незаращение неба («волчья пасть»). Также назначаются медикаментозные препараты для стимуляции опорожнения кишечника и отхода кишечных газов, так как эти процессы затруднены в силу патологий кишечника.
Дети в течение жизни страдают от синуситов, конъюнктивитов, отитов, пневмонии, инфекций мочеполовой системы. Часто у них развивается рак почки. Лечение в таких случаях симптоматическое.
Источник