
Элерса данлоса синдром мкб 10

Содержание
- Описание
- Дополнительные факты
- Симптомы
- Причины
- Классификация
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Элерса – Данлоса.
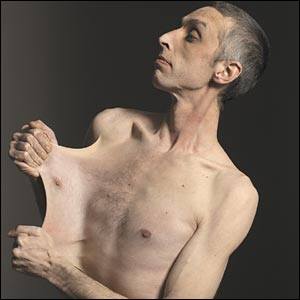
Гиперрастяжимость кожи при синдроме Элерса — Данлоса
Описание
Синдром Элерса. Данлоса – наследственная системная соединительнотканная дисплазия, обусловленная недостаточным развитием коллагеновых структур. В зависимости от клинического типа синдром Элерса-Данлоса может проявляться гипермобильностью суставов, необычайной ранимостью и растяжимостью кожи, склонностью к кровоизлияниям и кровотечениям, деформациями позвоночника и грудной клетки, миопией, косоглазием, птозом внутренних органов и пр. При диагностике синдрома Элерса-Данлоса учитываются клинические данные, результаты биопсии кожи и генотипирования; возможна пренатальная диагностика патологии. Лечение синдрома Элерса-Данлоса сводится к соблюдению щадящего режима, белковой диеты, симптоматической терапии.
Дополнительные факты
Синдром Элерса-Данлоса (несовершенный десмогенез, гиперэластическая кожа), наряду с несовершенным остеогенезом, синдромом Марфана и другими заболеваниями, относится к наследственным коллагенопатиям. Синдром Элерса-Данлоса неоднороден и включает в себя гетерогенную группу наследственных поражений соединительной ткани (соединительнотканных дисплазий), связанных с нарушением биосинтеза белка коллагена. Проявления синдрома Элерса-Данлоса носят системный характер и затрагивают опорно-двигательный аппарат, кожу, сердечно-сосудистую, зрительную, зубочелюстную и другие системы. Поэтому синдром Элерса-Данлоса представляет практический интерес не только для генетики, но и травматологии и ортопедии, дерматологии, кардиологии, офтальмологии, стоматологии.
Сложность верификации и наличие легких форм затрудняет получение точных сведений об истинной распространенности синдрома Элерса-Данлоса; частота диагностированных среднетяжелых случаев составляет 1:5 000 новорожденным, тяжелых форм – 1:100 000.
Гипермобильность суставов при синдроме Элерса — Данлоса
Симптомы
Ассоциированные симптомы: Боль в костях таза.
Причины
Различные варианты синдрома Элерса-Данлоса различаются по типу наследования, первичным молекулярным и биохимическим дефектам. Однако в основе всех клинических форм лежат мутации генов, обусловливающие количественную или структурную патологию коллагена. На сегодняшний день молекулярные механизмы синдрома Элерса-Данлоса установлены не для всех форм заболевания.
Так, известно, что I тип синдрома характеризуется снижением активности фибробластов, усилением синтеза протеогликанов, отсутствием ферментов, отвечающих за нормальный биосинтез коллагена. Синдром Элерса-Данлоса IV типа связан с недостаточностью продукции коллагена III типа; при VI типе заболевания имеет место недостаточность фермента лизилгидроксилазы, участвующего в гидроксилировании лизина в молекулах проколлагена. VII тип обусловлен нарушением превращения проколлагена I типа в коллаген; X тип – патологией плазменного фибронектина, участвующего в организации межклеточного матрикса.
Патоморфологическая картина при различных типах синдрома Элерса-Данлоса характеризуется истончением дермы, нарушением ориентации и потерей компактности коллагеновых волокон, разрастанием эластических волокон, увеличением числа сосудов и расширением их просвета.
Классификация
Всего выделяют 10 типов синдрома Элерса-Данлоса, различающихся по генетическому дефекту, характеру наследования и клиническим проявлениям. Рассмотрим основные из них:
I тип синдрома Элерса. Данлоса (классический тяжелого течения) – наиболее частый вариант заболевания (43% случаев) с аутосомно – доминантным типом наследования. Ведущим симптомом является гиперэластичность кожи, растяжимость которой по сравнению с нормой увеличена в 2-2,5 раза. Характерна гипермобильность суставов, носящая генерализованный характер, деформации скелета, повышенная ранимость кожи, склонность к наружным кровотечениям, образованию рубцов, плохому заживлению ран. У части больных выявляется наличие моллюскоподобных псевдоопухолей и варикозного расширения вен нижних конечностей. Беременность у женщин с I типом синдрома Элерса-Данлоса часто осложняется преждевременными родами.
II тип синдрома Элерса. Данлоса (классический мягкого течения) – характеризуется вышеописанными признаками, но выраженными в меньшей степени. Растяжимость кожи превосходит нормальную лишь на 30%; гипермобильность отмечается преимущественно в суставах стоп и кистей; кровоточивость и наклонность к рубцеванию незначительны.
III тип синдрома Элерса. Данлоса – имеет аутосомно – доминантное наследование, доброкачественное течение. Клинические проявления включают генерализованную повышенную подвижность суставов, скелетно-мышечные деформации. Остальные проявления (гиперэластичность и рубцевание кожи, геморрагии) минимальны.
IV тип синдрома Элерса. Данлоса – встречается редко, протекает тяжело; может наследоваться различными путями (доминантно или рецессивно). Гиперэластичность кожи незначительна, отмечается повышенная подвижность только суставов пальцев рук. Ведущим проявлением данного типа заболевания является геморрагический синдром: склонность к образованию экхимозов, спонтанных гематом (в т. Во внутренних органах), разрывам полых органов и сосудов (в т. Аорты). Сопровождается высокой летальностью.
V тип синдрома Элерса. Данлоса – имеет Х – сцепленное рецессивное наследование. Характеризуется повышенной растяжимостью кожи, умеренно выраженными гипермобильностью суставов, кровоточивостью и ранимостью кожи.
VI тип синдрома Элерса. Данлоса – наследуется по аутосомно – рецессивному типу. Кроме гиперэластичности кожи, наклонности к кровотечениям, повышенной подвижности суставов, имеются мышечная гипотония, тяжелый кифосколиоз, косолапость. Характерной чертой синдрома Элерса-Данлоса VI типа является глазной синдром, проявляющийся близорукостью, кератоконусом, косоглазием, глаукомой, отслойкой сетчатки.
VII тип синдрома Элерса. Данлоса (артроклазия) – наследуется как аутосомно – доминантно, так и аутосомно – рецессивно. Клиническую картину определяет низкий рост пациентов и гиперподвижность суставов, приводящая к частым привычным вывихам.
VIII тип синдрома Элерса. Данлоса – преимущественно наследуется аутосомно – доминантно. Ведущую роль в клинике играет хрупкость кожи, выраженный периодонтит, приводящий к ранней потере зубов.
X тип синдрома Элерса. Данлоса – характеризуется аутосомно – рецессивным наследованием; умеренной гиперэластичностью кожи и гипермобильностью суставов, стриями (полосовидной атрофией кожи), нарушением агрегации тромбоцитов.
XI тип синдрома Элерса. Данлоса – имеет аутосомно – доминантный тип наследования. У больных отмечаются рецидивирующие вывихи плечевых суставов, вывихи надколенника, встречается врожденный вывих бедра.
IX тип (Х-спепленный вариант вялой кожи) в настоящее время исключен из классификации синдрома Элерса-Данлоса. В современном варианте классификации синдрома Элерса-Данлоса рассматривается 7 основных типов заболевания:
• классический (типы I и II).
• гипермобильный (тип III).
• сосудистый (тип IV).
• кифосколиоз (тип VI).
• артроклазия (тип VIIB).
• дермоспараксис (тип VIIC).
• недостаток тенасцина-X.
Диагностика
Диагностика синдром Элерса-Данлоса проводится медицинским генетиком на основании генеалогических данных, анамнеза, клинического анализа, молекулярно-генетических исследований. Предварительно синдром Элерса-Данлоса может быть заподозрен при наличии больших диагностических критериев (гипермобильности суставов, гиперэластичности кожи, склонности к кровотечениям) и дополнительных малых (хрупкости кожи, патологии сердца, сосудов, глаз ).
Некоторые формы заболевания требуют проведения биопсии кожи для гистологического, гистохимического, электронно-микроскопического исследования.
Наличие в семье больных синдромом Элерса-Данлоса является показанием к медико-генетическому консультировании и проведению инвазивной пренатальной диагностики.
Больные с различными типами синдром Элерса-Данлоса могут нуждаться в наблюдении и обследовании детским травматологом-ортопедом, детским кардиологом, детским офтальмологом, детским стоматологом, сосудистым хирургом.
Лечение
Эффективная специфическая терапия синдрома Элерса-Данлоса не разработана. Детям требуется создание щадящего режима, исключающего излишнюю травматизацию суставов и кожи; ограничение физических нагрузок; соблюдение белковой диеты с включением в рацион костных бульонов, заливных блюд, студня. Обязательны регулярные курсы массажа, лечебной физкультуры, физиотерапии (магнитотерапии, электрофореза, лазеропунктуры).
Медикаментозная терапия синдрома Элерса-Данлоса включает применение аминокислот (карнитина), витаминов (С, Е, D, группы В), хондроитина сульфата, глюкозамина, минеральных комплексов (препаратов кальция и магния), метаболических препаратов (рибоксин, АТФ, коэнзим Q10) повторными курсами до1-1,5 мес. 2-3 раза в год.
При синдроме Элерса-Данлоса может быть показано хирургическое лечение: реконструкция грудной стенки, удаление псевдоопухолей, коррекция ВПС и пр.
Прогноз
На качество и продолжительность жизни больных синдромом Элерса-Данлоса влияет тип заболевания. Наиболее серьезный прогноз имеет IV тип синдрома Элерса-Данлоса – летальный исход может наступить вследствие разрывов сосудов, внутренних органов и кровотечений. Наличие синдрома I типа существенно ограничивает качество жизни. Относительно благоприятно протекание II—III типов болезни.
В целом, наличие синдрома Элерса-Данлоса сопряжено со множеством социальных трудностей, ограничивает полноценную физическую активность и выбор профессии.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: Q79.6
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q65-Q79 Врожденные аномалии пороки развития и деформации костно-мышечной системы / Q79 Врожденные аномалии (пороки развития) костно-мышечной системы, не классифицированные в других рубриках
Определение и общие сведения[править]
Синдром Элерса-Данло (Ehlers-Danlos) – генетически гетерогенное заболевание, обусловленное разнообразными мутациями в генах коллагена, либо в генах, отвечающих за синтез ферментов, принимающих участие в созревании волокон коллагена. Характеризуется гиперэластичностью кожи, подкожными сферулами, переразгибанием суставов, лёгкой ранимостью тканей и геморрагическим синдромом.
Эпидемиология
Истинная распространённость неизвестна вследствие сложности верификации и большого числа лёгких форм, частота диагностированных случаев – 1 на 5000 новорождённых, тяжёлые формы встречают редко (1:100 000).
Классификация
В генетической классификации описано 11 типов и ряд подтипов с разным наследованием, клиническими особенностями и биохимическими дефектами.
Этиология и патогенез[править]
Синдром Элерса-Данло – группа заболеваний соединительной ткани, различающихся по типу наследования, клиническим особенностям и биохимическому дефекту. В большинстве случаев наследуется по аутосомно-доминантному типу, сопровождается уменьшением количества или изменением структуры коллагена.
Клинические проявления[править]
Синдром Элерса-Данло: Диагностика[править]
Объём обследования определяется наличием ведущих клинических признаков заболевания. Существенное значение имеет генеалогическое исследование и молекулярно-генетические методы диагностики.
Диагностику СЭД проводят согласно Вилльфраншской классификации (Beighton P. et аl., 1998).
Для диагностики СЭД необходимо выполнение следующих требований.
• Для клинической диагностики необходимо наличие хотя бы одного большого критерия. При соответствующих возможностях наличие одного или более больших критериев гарантирует подтверждение СЭД на лабораторном уровне.
• Малый критерий – признак, обладающий меньшим уровнем диагностической специфичности. Наличие одного или более малых критериев вносит свой вклад в диагностику того или иного типа СЭД.
• При отсутствии больших критериев для установления диагноза малых недостаточно. Наличие малых критериев даёт основание предполагать наличие состояния, подобного СЭД, характер которого будет разъясняться по мере того, как станет известной его молекулярная основа. Поскольку встречаемость малых критериев существенно выше, чем больших, в полном согласии с Вилльфраншским пересмотром наличие только малых критериев даёт основание для диагностики элерсоподобного фенотипа.
Критерии диагностики СМ и СЭД включают гипермобильность суставов.
В случае невыполнения соответствующих критериев гипермобильность необходимо рассматривать как самостоятельное состояние.
Дифференциальный диагноз[править]
Синдром Элерса-Данло: Лечение[править]
Принципы лечения
Богатая белком диета, содержащая костные бульоны, студни, заливные блюда. Курсы массажа, физиотерапии, лечебная физкультура. Посиндромная терапия, зависящая от выраженности органных изменений. Медикаментозное лечение с использованием аминокислотных (карнитин, витаминных (витамины D, С, Е, В1, В2, В6), минеральных комплексов (кальция карбонат/холекальциферол, магнерот), хондроитина сульфата перорально и местно, глюкозамин, оссеин-гидроаппатитных комплексов (остеокеа, остеогенон), трофических препаратов (АТФ, инозин, лецитин, коэнзим Q10). Указанные препараты принимают сочетанными курсами 2-3 раза в год продолжительностью 1-1,5 мес
Профилактика[править]
Прочее[править]
Прогноз
Прогноз благоприятный, серьёзнее при I (вследствие артропатий) и IV (вследствие кровотечений и разрывов сосудов) типах СЭД. Детей следует ориентировать на выбор профессии, не связанной с физическими нагрузками, работой стоя.
Источники (ссылки)[править]
Педиатрия [Электронный ресурс] / Под ред. А.А. Баранова – М. : ГЭОТАР-Медиа, 2009. – https://www.rosmedlib.ru/book/ISBN9785970410851.html
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Элерса-Данло (Ehlers-Danlos) (СЭД; Q79.6) – генетически гетерогенное заболевание, обусловленное разнообразными мутациями в генах коллагена, либо в генах, отвечающих за синтез ферментов, принимающих участие в созревании волокон коллагена.
Эпидемиология
Истинная распространённость неизвестна вследствие сложности верификации и большого числа лёгких форм. Распространенность cEDS была оценена в 1:20 000 [Byers 2001]. Однако, вероятно, что некоторые люди с более легкими проявлениями заболевания, ранее классифицированные как EDS типа II, не обращаются к врачу и, следовательно, остаются незамеченными.
Причины синдрома Злерса-Данло
Синдром Элерса-Данло – группа заболеваний соединительной ткани, различающихся по типу наследования, клиническим особенностям и биохимическому дефекту. В большинстве случаев наследуется по аутосомно-доминантному типу, сопровождается уменьшением количества или изменением структуры коллагена. Описана связь дефицит белка Tenascin-X с риском развития синдромом Элерса-Данлоса. [1]
Описано 2 основных способа наследования синдрома Элерса-Данло:
- аутосомно-доминантное наследование (гипермобильная, классическая и сосудистая EDS) – дефектный ген, который вызывает EDS, передается одним родителем, и риск развития этого заболевания у каждого их детей составляет 50%
- аутосомно-рецессивное наследование (кифосколиотическая EDS) – дефектный ген унаследован от обоих родителей, и риск развития этого заболевания у каждого их детей составляет 25%
Человек с синдромом Элерса-Данло может передавать детям только один тип синдрома.
Например, дети человека с гипермобильной EDS не могут наследовать сосудистуюEDS.
Тяжесть состояния может варьироваться в пределах одной семьи. [2]
Патогенез
Изучение этих заболеваний позволило по-новому взглянуть на молекулярный патогенез EDS, вовлекая генетические дефекты в биосинтез других молекул внеклеточного матрикса (ECM), таких как протеогликаны и тенасцин-X, или генетические дефекты в молекулах, секреция и сборка белков ЕСМ. [3] При сосудистом типе EDS были идентифицированы мутации в коллагене типа III (EDS IV) (Kuivaniemi et al. 1997). Структурные мутации, влияющие на расщепление N-протеиназой проколлагена I, были обнаружены в редких вариантах EDS (EDS VII A и B) (Byers et al. 1997). [4]
В настоящее время считается, что примерно 50% пациентов с клиническим диагнозом классического синдрома Элерса-Данлоса имеют мутации в генах COL5A1 и COL5A2, кодирующие α1 и α2-цепь коллагена типа V соответственно. [5]
Симптомы синдрома Злерса-Данло
Характеризуется гиперэластичностью кожи, подкожными сферулами, переразгибанием суставов, лёгкой ранимостью тканей и геморрагическим синдромом. [6]
Кожа хрупкая, что проявляется в наличием рубцов и ран после относительно незначительной травмы, особенно над точками давления (колени, локти) и участками, подверженными травме (голени, лоб, подбородок). Заживление ран слабое. Шрамы становятся широкими, с «папиросной» (папирусной) внешностью.
Другие дерматологические особенности в cEDS:
- Моллюскоидные псевдотуморы.
- Подкожные сфероиды.
- Пьезогенные папулы: маленькие, болезненные, обратимые грыжи нижележащих глобул жировой ткани через фасцию в дерму, например, на медиальной и латеральной сторонах стоп при стоянии.
- Elastosis perforans serpiginosa: редкое заболевание кожи неизвестной этиологии, характеризующееся красными или эритематозными кератотическими папулами, некоторые увеличиваются наружу в серпигинусной или дугообразной конфигурации, оставляя слегка атрофические очаги.
- Акроцианоз: безболезненное заболевание, вызванное сужением или сужением мелких кровеносных сосудов кожи (затрагивающее главным образом руки), при котором пораженные участки становятся синими и становятся холодными и потными; может возникнуть локальный отек.
- Озноб: травмы от холода, характеризующиеся красной опухшей кожей, нежной и горячей на ощупь, которая может зудеть; может развиться менее чем за два часа на коже, подвергшейся воздействию холода.
Проявления генерализованной растяжимости и ломкости тканей наблюдаются во многих органах:
- Цервикальная недостаточность во время беременности.
- Паховая и пупочная грыжа.
- Хиатальная и послеоперационная грыжа.
- Рецидив выпадения прямой кишки в раннем детстве.
Суставы
- Осложнения гипермобильности суставов, включая вывихи плеча, надколенника, пальцев, бедра, радиуса и ключицы, могут возникать и обычно проходят спонтанно или легко управляются больным человеком. Некоторые люди с cEDS могут испытывать хронические боли в суставах и конечностях, несмотря на нормальную рентгенограмму скелета.
Другие особенности включают гипотонию с задержкой моторного развития, усталость и мышечные спазмы, а также легкие кровоподтеки. Пролапс митрального клапана может встречаться нечасто.
Формы
Синдромы Элерса-Данлоса включают гетерогенную группу заболеваний, которые характеризуются хрупкостью мягких соединительных тканей и широко распространенными проявлениями в коже, связках и суставах, кровеносных сосудах и внутренних органах. Клинический спектр варьируется от мягкой кожной и суставной гиперлаксии до тяжелой физической инвалидности и опасных для жизни сосудистых осложнений.
Первоначально 11 форм синдрома Элерса-Данлоса были названы римскими цифрами для обозначения типов (тип I, тип II и т. Д.). В 1997 году исследователи предложили более простую классификацию (номенклатура Вильфранша), которая сократила количество типов до шести и дала им описательные названия на основе их основных характеристик. [7]
Текущая классификация Villefranche распознает шесть подтипов, большинство из которых связаны с мутациями в одном из генов, кодирующих фибриллярные белки коллагена или ферменты, участвующие в посттрансляционной модификации этих белков. [8]
- Тип I Классический тип (OMIM 606408)
- Тип II Классический тип, Синдром Элерса-Данлоса с дефицитом Тенасцина Х
- Тип III Тип гипермобильности
- Тип VIA, Тип VIB Сосудистый тип (OMIM 225320)
- Типы VIIA и VIIB Тип артрохалазии (OMIM 130060, 617821), Тип VIIC Дерматоспараксис (OMIM 225410), Тип прогероида
- Тип VIII Тип пародонтита, Вариант Элерса-Данлоса с перивентрикулярной гетеротопией
Установление правильного подтипа EDS имеет важные последствия для генетического консультирования и управления и поддерживается конкретными биохимическими и молекулярными исследованиями. [9]
Диагностика синдрома Злерса-Данло
Объём обследования определяется наличием ведущих клинических признаков заболевания. Существенное значение имеет генеалогическое исследование и молекулярно-генетические методы диагностики.
Для диагностики синдрома Элерса-Данло необходимо выполнение следующих требований.
- Для клинической диагностики необходимо наличие хотя бы одного большого критерия. При соответствующих возможностях наличие одного или более больших критериев гарантирует подтверждение синдрома Элерса-Данло на лабораторном уровне.
- Малый критерий – признак, обладающий меньшим уровнем диагностической специфичности. Наличие одного или более малых критериев вносит свой вклад в диагностику того или иного типа синдрома Элерса-Данло.
- При отсутствии больших критериев для установления диагноза малых недостаточно. Наличие малых критериев даёт основание предполагать наличие состояния, подобного синдрома Элерса-Данло, характер которого будет разъясняться по мере того, как станет известной его молекулярная основа. Поскольку встречаемость малых критериев существенно выше, чем больших, в полном согласии с Вилльфраншским пересмотром наличие только малых критериев даёт основание для диагностики элерсоподобного фенотипа.
Диагноз классического синдрома устанавливается у пациента на основе минимальных клинико – диагностических критериев (гиперэластичность кожи и наличие атрофических рубцов) и идентификации на молекулярно-генетическом тестировании в патогенного гена COL5A1, COL5A2 или COL1A1.
Критерии диагностики синдрома Морфана и синдрома Элерса-Данло включают гипермобильность суставов. В случае невыполнения соответствующих критериев гипермобильность необходимо рассматривать как самостоятельное состояние.
Какие анализы необходимы?
Лечение синдрома Злерса-Данло
Междисциплинарная программа реабилитации, сочетающая физическую и когнитивно-поведенческую терапию показала значительные изменения в восприятии повседневной деятельности, значительное увеличение мышечной силы и выносливости и значительное снижение кинезиофобии. Произошли меньшие изменения в ощущаемой боли. Участники также сообщили об увеличении участия в повседневной жизни.
Богатая белком диета, содержащая костные бульоны, студни, заливные блюда. Курсы массажа, физиотерапии, лечебная физкультура. [10] Посиндромная терапия, зависящая от выраженности органных изменений. Медикаментозное лечение с использованием аминокислотных (карнитин, нутраминос), витаминных (витамины D, С, Е, В1, В2, В6), минеральных комплексов (магнеВ кальций-D3-Никомед, магнерот), хондроитина сульфата перорально и местно, глюкозамин, оссеин-гидроаппатитных комплексов (остеокеа, остеогенон), трофических препаратов (АТФ, инозин, лецитин, коэнзим Q10). Указанные препараты принимают сочетанными курсами 2-3 раза в год продолжительностью 1-1,5 мес.
Прогноз
Синдром Элерса-Данлоса типа IV (EDS) является тяжелой формой. Пациенты часто имеют маленькую продолжительность жизни из-за спонтанного разрыва артерии большого калибра (например, селезеночной артерии, аорты) или перфорации внутренних органов. Артериальные аневризмы, пролапс клапанов и спонтанный пневмоторакс являются распространенными осложнениями. Прогноз с этим типом плохой. [11]
Другие типы, как правило, не так опасны, и люди с данным диагнозом могут вести здоровый образ жизни. Тип VI также несколько опасен, хотя встречается редко.
Детей следует ориентировать на выбор профессии, не связанной с физическими нагрузками, работой стоя.
Источник