
Какая болезнь человека результат генной мутации синдром

В настоящее время важное место в сфере медицинских наук занимает медицинская генетика, которая изучает генетические заболевания. Вы уже знаете, что
вариантов мутаций множество: от выпадения отдельных нуклеотидов в гене до утраты целых хромосом. Количество вариантов мутаций и их сочетаний – бесчисленно, что делает медицинскую генетику неисчерпаемой.
Медицинская генетика играет важную роль при планировании семьи, служит для предупреждения наследственных заболеваний. В данной статье мы изучим некоторые наиболее известные наследственные заболевания.

Альбинизм
Альбинизм (лат. albus — белый) – врожденное заболевание, наследуемое по рецессивному типу и связанное с нарушением синтеза
черного пигмента – меланина (греч. melanos – черный) у животных или хлорофилла (у растений). Альбинизм возникает в результате
генной мутации в участке ДНК, ответственном за синтез меланина/хлорофилла.
Растения с утратой хлорофилла утрачивают способность улавливать солнечный свет, поэтому полный альбинизм для них
заканчивается летально. У животных мутация происходит в гене тирозиназы, в связи с чем меланин не синтезируется: кожа
альбиносов не способна загорать, для них характерен больший риск ожогов и рака кожи.
Радужка пропускает свет и становится красноватого оттенка, за счет кровеносных сосудов, расположенных на глазном дне.
Серповидно-клеточная анемия
Это наследственное заболевание, вызванное генной мутацией, в результате которой меняется конформация молекулы гемоглобина:
эритроцит становится выгнутым и напоминает серп.
Эта болезнь встречается особенно часто в странах, эндемичных по малярии. Больные серповидно-клеточной анемией обладают
повышенной устойчивостью к заражению малярийным плазмодием, поэтому эту болезнь можно рассматривать как результат действия
естественного отбора: с ней выживаемость людей повышалась, и они продолжали род, передавая мутацию потомкам.

Синдром Дауна
Наследственное заболевание, возникающее в результате геномной патологии: трисомия по 21-ой паре хромосом. Это означает, что
вместо двух хромосом в 21-ой паре появляется одна лишняя – третья хромосома. Причина ее появления связана с нерасхождением
хромосом во время мейоза.
Риск рождения ребенка с синдромом Дауна возрастает с увеличением возраста матери.

Синдром проявляется характерными признаками: плоское лицо, приоткрытый рот, поперечная ладонная складка, гиперподвижность суставов,
эпикантус (кожная складка, прикрывающая угол глазной щели).

Синдром Эдвардса
Наследственное заболевание, вызванное геномной мутацией – трисомией по 18 паре хромосом. Причина – нерасхождение хромосом во время
мейоза, еще до оплодотворения. Чаще болезнь встречается у пожилых матерей.
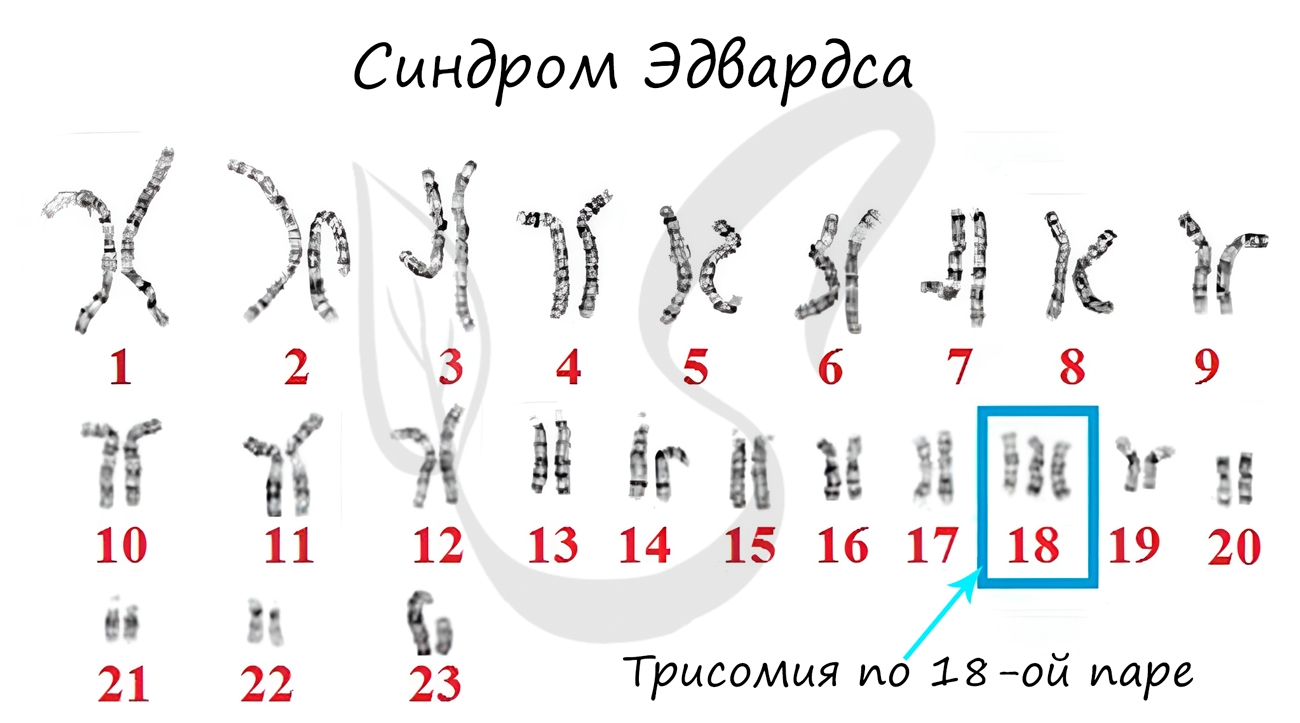
Детям с синдромом Эдвардса сопутствуют пороки сердца и сосудов: 60% детей умирают в течение первых 3 месяцев, до 1 года доживают лишь 5-10% детей.
Синдром Патау
Наследственное заболевание, обусловленной геномной мутацией – трисомией по 13 паре хромосом. Существует зависимость между возрастом матери
и вероятностью рождения ребенка с синдромом Патау (с возрастом риск увеличивается), хотя зависимость менее выражена, чем в случае с синдромом
Дауна.
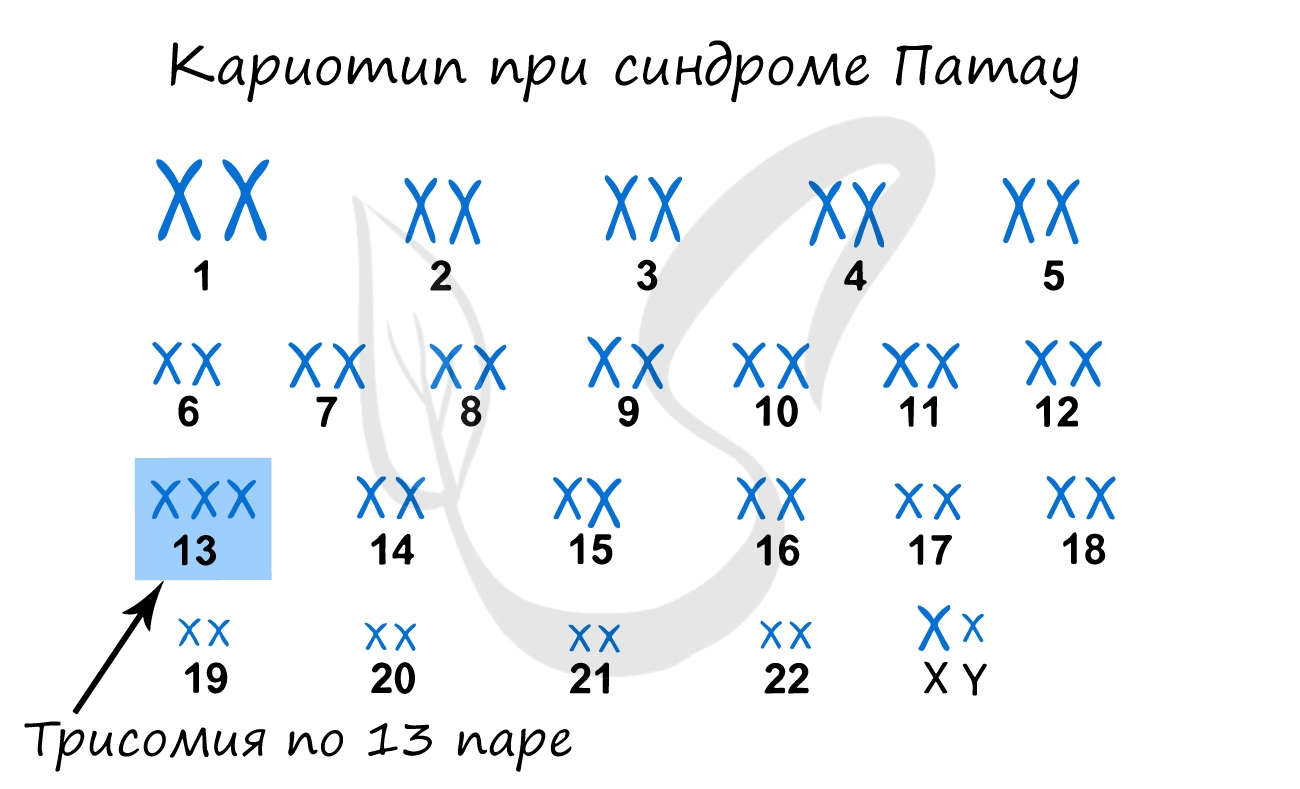
При данном синдроме обнаруживаются тяжелые врожденные пороки сердца и сосудов, нервной системы. Большинство детей с синдромом Патау умирают
в первые недели или месяцы жизни, до 1 года доживают лишь 5% детей.

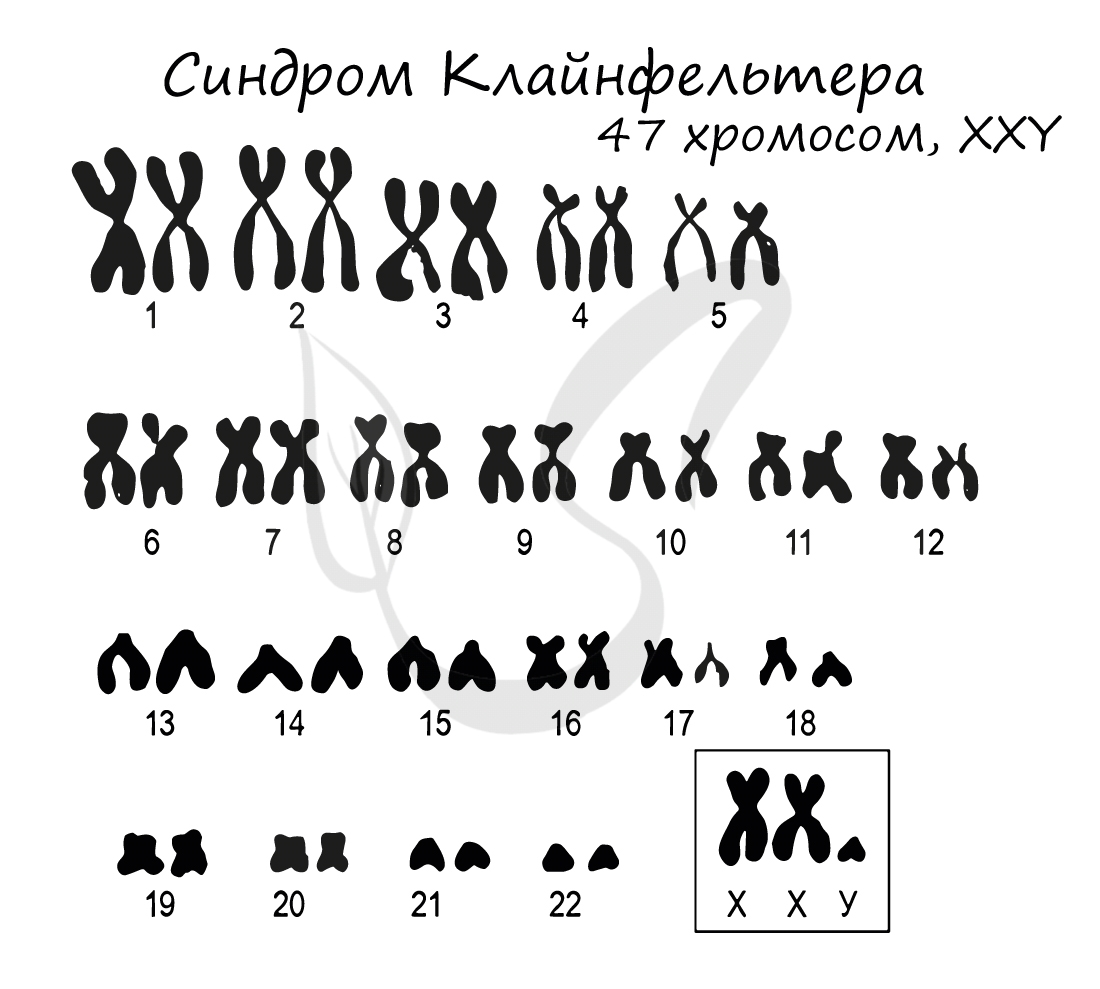
Синдром Клайнфельтера
Синдром Клайнфельтера представляет собой наследственное заболевание, развивающееся вследствие полисомии по X и Y хромосомам (половым
хромосомам). Возможны несколько вариантов генотипов: XXY (самый частый), XYY, XXXY, XXXXY, XXXYY. На всякий случай напомню норму
мужского генотипа – “XY” 🙂
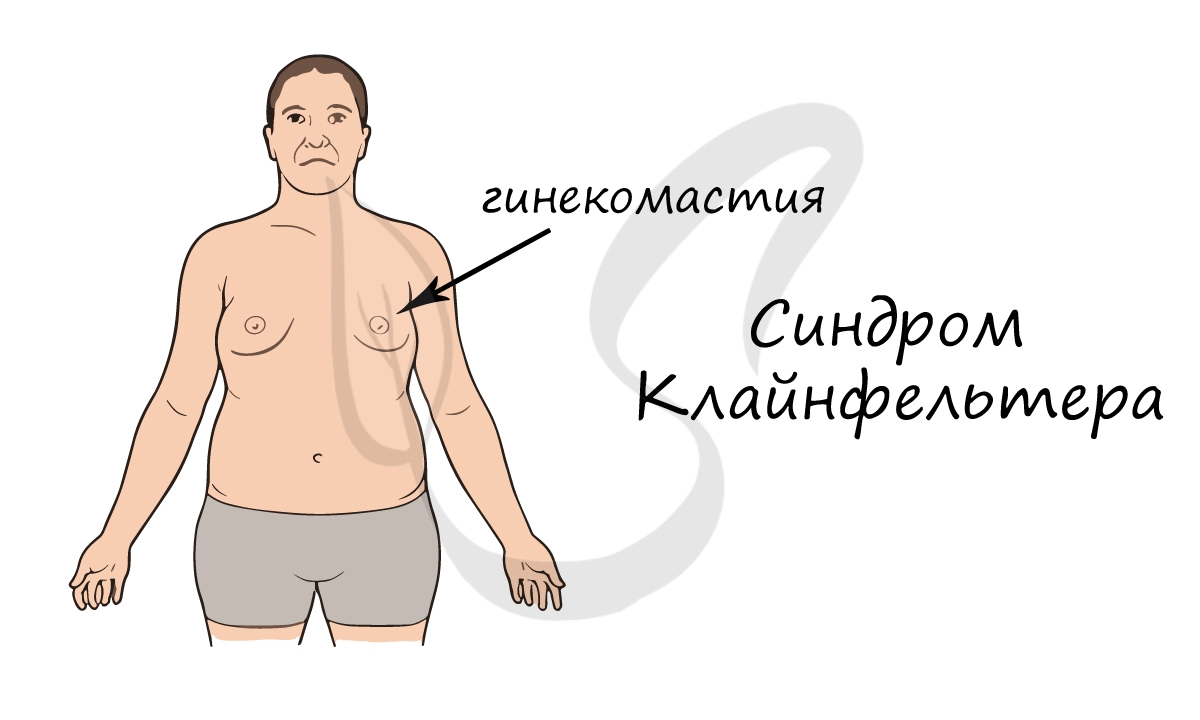
Диагностируется синдром относительно поздно, так как проявляется только после полового созревания. В подростковом возрасте развивается гинекомастия
(увеличение грудной железы), сохраняющаяся всю жизнь. Характерно наличие маленьких плотных яичек. Синдром Клайнфельтера приводит к бесплодию.
Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера – наследственное заболевание, характерное только для женщин и возникающее в результате моносомии по половым
хромосомам. Генотип человека при таком заболевании будет записан как X0 (45 хромосом).
Больные синдромом Шерешевского-Тернера низкорослые, инфантильные, их психический статус характеризуется состоянием беспричинно приподнятого
настроения – эйфорией. Тем не менее, интеллект и жизнеспособность сохранены. Из-за геномной мутации (X0) стерильны.

© Беллевич Юрий Сергеевич 2018-2021
Данная статья написана Беллевичем Юрием Сергеевичем и является его интеллектуальной собственностью. Копирование, распространение
(в том числе путем копирования на другие сайты и ресурсы в Интернете) или любое иное использование информации и объектов
без предварительного согласия правообладателя преследуется по закону. Для получения материалов статьи и разрешения их использования,
обратитесь, пожалуйста, к Беллевичу Юрию.
Источник
Синдром Ли – генетически гетерогенное наследственное заболевание, характеризующееся разнообразными нарушениями метаболизма и формирования компонентов центральной нервной системы. Симптомы этой патологии, как правило, проявляются еще в раннем детстве, к ним относят мышечную гипотонию, проблемы со вскармливанием и задержку психомоторного развития. При дальнейшем прогрессировании заболевания возникают эпилептические припадки, гиперкинезы, дыхательные расстройства. Диагностика синдрома Ли осуществляется на основании данных настоящего статуса больного, магнитно-резонансной томографии, молекулярно-генетических анализов. Специфического лечения данной патологии не существует, симптоматическая терапия лишь незначительно замедляет прогрессирование заболевания.
Общие сведения
Синдром Ли (подострая некротизирующая энцефаломиелопатия) – наследственное нейродегенеративное заболевание центральной нервной системы, которое характеризуется ранним началом и неуклонным прогрессированием неврологических нарушений. Впервые данное состояние было описано в 1951 году английским психиатром Денисом Ли, который определил его как наследственный вариант энцефаломиелопатии. Дальнейшие исследования показали, что синдром Ли является крайне гетерогенным состоянием с точки зрения этиологии – его причиной становятся дефекты множества генов, расположенных на аутосомах, Х-хромосоме и митохондриальной ДНК. По этой причине механизм наследования заболевания может быть (в зависимости от характера мутации) аутосомно-рецессивным, сцепленным с полом или митохондриальным. Из-за разнообразия генетических дефектов, являющихся причиной синдрома Ли, различается и половое распределение этого состояния, однако, по мнению многих врачей-генетиков, в целом можно считать, что оно в равной степени поражает как мальчиков, так и девочек. Встречаемость составляет ориентировочно 1 случай на 34-36 тысяч новорожденных.

Синдром Ли
Причины и классификация синдрома Ли
Причинами развития синдрома Ли могут выступать мутации широкого спектра генов, расположенных на разных хромосомах. Однако патогенез этого состояния примерно сходен у различных форм патологии и чаще всего связан с нарушением процессов клеточного дыхания и функционирования дыхательной цепи митохондрий. В отношении некоторых форм синдрома Ли также замечено нарушение функционирования пируватдегидрогеназного комплекса. Нарушение структуры белков дыхательной цепи митохондрий приводит к недостаточному синтезу АТФ, являющемуся основным источником энергии во всех клетках организма. Нейроны и клетки нейроглии особенно чувствительны к недостатку энергии, что становится причиной развития разнообразных нарушений еще с детского возраста. Классификация всех генетических дефектов при синдроме Ли основана на том, какой компонент дыхательной цепи (представляющей собой белковый комплекс) митохондрий нарушен в результате мутации.
- Синдром Ли, обусловленный поражением комплекса 1 (НАДН-KoQ-редуктаза). Этот вариант может наследоваться как аутосомно-рецессивно, так и митохондриально. Наиболее распространенные варианты заболевания этого типа обусловлены мутациями ядерных генов NDUFA10 (расположен на 19-й хромосоме), NDUFS4 и DUFAF2 (5-я хромосома), NDUFS3 (11-я хромосома). Кроме того, к развитию синдрома Ли в результате поражения митохондриального комплекса 1 способны приводить дефекты митохондриальной ДНК – генов MTND1, MTND2 и MTND3. Результатом этого является нарушение начального этапа переноса электронов и водорода в дыхательной цепи, что снижает синтез АТФ.
- Синдром Ли, вызванный дефектами белков, входящих в митохондриальный комплекс 2 (сукцинат-KoQ-редуктаза). Заболевание этого типа наследуется только аутосомно-рецессивно, достоверно удалось установить взаимосвязь между синдромом Ли и мутациями гена SDHA, локализованного на 5-й хромосоме. Данный ген кодирует субъединицу А сукцинатдегидрогеназного комплекса, при генетических нарушениях такого типа активность фермента снижается, что и ведет к развитию заболевания.
- Синдром Ли, возникающий в результате нарушения структуры белков митохондриального комплекса 3 (KoQН2-цитохром с-редуктаза). К этой разновидности относят наиболее распространенный вариант заболевания, вызванный мутацией гена BCS1L, расположенного на 2-й хромосоме. Развитие синдрома Ли обусловлено пониженной экспрессией фермента убихинон-с-редуктазы, входящего в состав митохондриального комплекса 3. Его выделение регулируется специфическим белком, который кодируется геном BCS1L – в результате мутации полученный дефектный протеин не способен полноценно выполнять свои функции. Для этого варианта синдрома Ли характерно аутосомно-рецессивное наследование.
- Синдром Ли, обусловленный повреждением митохондриального комплекса 4 (цитохром с-оксидаза). Может быть вызван как мутациями ядерных генов (COX10, SCO1), в основном расположенных на 17-й хромосоме, так и повреждением митохондриальной ДНК – это удалось выяснить по характеру наследования некоторых форм, однако ключевые гены пока не определены.
- Синдром Ли, вызванный нарушением структуры митохондриального комплекса 5 (АТФ-синтаза). К этому варианту относят сравнительно редкие мутации гена ATPAF2, локализованного на 17-й хромосоме. В результате мутации нарушается работа АТФ-синтазы, образование АТФ окислительным путем резко снижается.
В качестве отдельного варианта синдрома Ли часто указывают форму заболевания, обусловленную мутациями гена PDHA1, который расположен на Х-хромосоме. В результате наследование данного типа патологии является сцепленным с полом – болеют почти исключительно мальчики, тогда как женщины выступают носительницами патологических генов. Митохондриальный тип наследования синдрома Ли также имеет множество особенностей. Передача патологических генов происходит от матери к потомству и продолжается только по женской линии. Поскольку каждая митохондрия имеет собственную молекулу ДНК, в клетке одновременно присутствуют как «здоровые», так и «больные» органеллы, а при делении клеток (в том числе и при мейозе в процессе образования яйцеклеток) распределение больных генов оказывается неодинаковым. Женщины с относительно небольшим процентом «больных» митохондрий в клетках могут быть фенотипически здоровыми, но передавать их своему потомству. Невозможно точно предсказать, какое количество патологических митохондрий получит ребенок таких носителей, поэтому вероятность развития синдрома Ли у детей этих женщин неопределенная.
Симптомы
Проявления синдрома Ли обычно возникают на протяжении первого года жизни ребенка, иногда они могут регистрироваться в возрасте 2-5 лет, в редких случаях развитие заболевания начинается в подростковый период. Обычно первым проявлением патологии становится сонливость или, наоборот, повышенная возбудимость ребенка, у грудных детей наблюдается нарушение питания, недобор массы тела. В дальнейшем синдром Ли приводит к задержке психофизического развития, а у детей старшего возраста – к постепенной утрате уже обретенных навыков. Среди других неврологических симптомов заболевания наиболее часто отмечаются парезы, тремор конечностей, нарушение координации движения, поражение периферических нервов, снижение сухожильных рефлексов. В дальнейшем могут регистрироваться клонические судороги и эпилептические припадки.
Из-за недостатка энергии, обусловленного синдромом Ли, страдает не только нервная система, но и другие органы с высоким потреблением АТФ. В большинстве случаев у больных детей отмечается мышечная гипотония и слабость. Затрагивает заболевание и печень – орган с очень высоким потреблением энергии. У пациентов с синдромом Ли нередко выявляется увеличение печени, желтуха, иногда гепатолиенальный синдром. По мере прогрессирования патологии возникают нарушения дыхания – оно становится затрудненным, иногда приобретает характер дыхания Чейна-Стокса. У ряда больных со временем развивается миокардиодистрофия.
Синдром Ли имеет прогрессирующие течение. На терминальных этапах наблюдается поражение органов зрения, которое проявляется нистагмом, нарушением цветовосприятия, косоглазием. В конечном итоге может возникнуть атрофия зрительного нерва и полная слепота. Мышечная гипотония и гипорефлексия сменяются спастическим напряжением мышц и повышением сухожильных рефлексов. Через 2-7 лет после появления первых симптомов синдрома Ли происходит резкое падение массы тела, вышеперечисленные проявления резко усиливаются, наступает летальный исход по причине дыхательной или сердечно-сосудистой недостаточности.
Диагностика и лечение синдрома Ли
Для определения наличия синдрома Ли применяют магнитно-резонансную томографию головного мозга, электронейромиографию, изучение наследственного анамнеза, молекулярно-генетические анализы. При осмотре выявляют характерные неврологические симптомы, тремор конечностей, отставание в психофизическом развитии, у младенцев – недобор массы тела. На магнитно-резонансной томографии мозга обнаруживают симметричные изменения в области продолговатого мозга, таламуса и моста, иногда аналогичные изменения могут регистрироваться и в спинном мозге. Наилучшие результаты диагностики синдрома Ли при помощи МРТ получаются при использовании T2W и FLAIR режимов.
В тех случаях, когда имеются признаки поражения периферических нервов и мышц, для диагностики синдрома Ли выполняют электронейромиографию. При этом заболевании главным и наиболее частым результатом ЭНМР становится замедление скорости прохождения нервного импульса, которое свидетельствует о демиелинизации нервов. Изучение наследственного анамнеза информативно в случае аутосомно-рецессивных форм заболевания, при мутации генов митохондриальной ДНК четко определить семейный характер патологии затруднительно. Молекулярно-генетическая диагностика массово используется только в отношении некоторых форм синдрома Ли (обусловленных мутациями генов BCS1L, SURF1 и некоторых других).
Специфического лечения данной патологии не существует, применяется симптоматическая терапия: противосудорожные и ноотропные средства, препараты для улучшения мозгового кровообращения. Важную роль в лечении синдрома Ли играет назначение витаминов, служащих кофакторами ферментов дыхательной цепи митохондрий – В1, В6, Q10. Их регулярный прием позволяет несколько замедлить прогрессирование заболевания и уменьшить выраженность симптомов. Однако, несмотря на все предпринятые терапевтические меры, 80% больных умирает через 2-7 лет после регистрации первых проявлений патологии.
Прогноз и профилактика
Прогноз синдрома Ли крайне неблагоприятный, так как большинство больных умирает через несколько лет после возникновения заболевания. Симптоматическое лечение может несколько замедлить прогрессирование патологии и ослабить выраженность проявлений, однако полноценного улучшения оно не обеспечивает. В большинстве случаев еще за год-два до летального исхода наступает полная инвалидизация больного, обусловленная неврологическими, дыхательными и метаболическими нарушениями. Причиной смерти при синдроме Ли чаще всего становится сердечно-сосудистая или дыхательная недостаточность. Профилактика этого заболевания осуществляется в рамках медико-генетического консультирования родителей перед зачатием ребенка.
Источник
Преимплантационная генетическая диагностика — возможность узнать о возможных рисках наследственных заболеваний.
Услуги и цены…
Узнать о предрасположенности к наследственным патологиям можно с помощью идентификации генов.
Подробнее об услуге…
Если в семье имелись случаи заболевания сахарным диабетом, имеет смысл воспользоваться услугой идентификации генов.
Подробнее об услуге…
Лекарственные средства могут повлиять на результаты идентификации генов методом полимеразной цепной реакции.
Подробнее…
«Это у нас в семье наследственное», — мы часто говорим так по отношению к самым разным вещам. Под понятие «наследственное» может попадать и цвет волос, и телосложение, и постоянные простуды. Особенно часто мы оправдываемся наследственностью, имея в виду болезни, что далеко не всегда соответствует действительности. Что же собой представляют генетические, или наследственные, заболевания, как их диагностируют и можно ли их предотвратить?
Что такое генетические болезни? Обременительное наследство
Для начала необходимо разобраться в терминах. Начнем с того, что генетические заболевания и заболевания, к которым выявлена наследственная предрасположенность, — разные понятия.
- Генетические болезни обусловлены нарушениями в строении генома (отсюда другое название — моногенные заболевания). В качестве примера можно привести галактоземию. При этом заболевании плохо работают ферменты, которые превращают молочный сахар в глюкозу. Уже выявлен ген, «отвечающий» за развитие заболевания. Более того, выяснено, что если ребенок получает «дефектный» ген от одного из родителей, то ферментная система работает примерно на 50%, а если от обоих, то всего на 10%[1].
- Заболевания, к которым у человека есть наследственная предрасположенность, зависят не только от генетики, но и от факторов внешней среды: того, где мы живем, сколько двигаемся, что едим. Например, у человека может быть склонность к атеросклерозу, но правильный образ жизни и рациональное питание помогают ему оставаться здоровым.
Чтобы понять принцип передачи наследственных заболеваний, надо вспомнить, что такое гены. Условно говоря, это некий набор «карт памяти», на каждой из которых «записаны» определенные данные об организме человека. Если же говорить научным языком, то ген — это фрагмент нашей ДНК. Совокупность генов (а их число доходит до 25 000[2]), представляющая собой плотно свернутую нить ДНК, — это хромосома. Всего у человека их 23 пары. Это весь наш генетический багаж, или иначе — геном.
Каждая из 23 хромосом имеет свою пару. Записанная в структуре одной хромосомы информация дублируется на парной. То есть любой признак, будь то цвет глаз или предрасположенность к сердечно-сосудистым заболеваниям, кодируется двумя генами. Они могут быть идентичными, но могут и отличаться (такие гены называют аллелями). Например, один из двух генов, определяющий цвет глаз, может «кодировать» серый оттенок, а второй — карий. Скорее всего, у носителя таких аллелей цвет глаз будет карий, так как ген, несущий эту информацию, является доминантным. Второй же ген (серый цвет глаз) более «слабый» — рецессивный[3].
Теперь разберемся в механизме наследования. Формируясь, зародыш получает половину хромосом от матери, а половину — от отца. Именно поэтому организм ребенка не копирует ни одного из родителей, а имеет свою индивидуальность. Передача хромосом, генов, а значит, и передача информации о наследственных заболеваниях, возможна по нескольким схемам:
- аутосомно-доминантный. Если ребенок получает «сильный», доминантный, ген хотя бы от одного из родителей, то этот ген обязательно проявится. Таким образом передается, например, ахондроплазия — заболевание, при котором нарушается рост конечностей, а кости становятся ломкими[4].
- аутосомно-рецессивный. Здесь чуть сложнее — признак проявляется только в том случае, если ребенок получил от родителей два «слабых», рецессивных, гена. Вероятность проявления заболевания ниже, чем в первом случае. Таким образом передаются по наследству фенилкетонурия, альбинизм и другие заболевания[5].
- кодоминантный. При этом типе наследования проявляются оба гена — и доминантный, и рецессивный. Примером может быть серповидно-клеточная анемия: наличие активных доминантного и рецессивного генов приводит к тому, что в крови обнаруживается и нормальная, и патологическая форма гемоглобина.
- наследование, сцепленное с полом. Известно, что половые хромосомы у мужчин и женщин различаются: у женщин две Х-хромосомы, а у мужчины — X и Y. К половым хромосомам «привязаны» некоторые важные признаки и информация о заболеваниях. Например, гемофилией, как известно, болеют почти исключительно мужчины[6]: если в Х-хромосоме у мужчин содержится ген, отвечающий за патологию, то Y-хромосома никак его не компенсирует, там этого гена нет[7]. По этому же принципу передаются дальтонизм, мышечная дистрофия Дюшена и т.д.
К наиболее распространенным генетическим заболеваниям относятся:
- дальтонизм — около 850 случаев на 10 000;
- расщепление позвоночника — 10–20 случаев на 10 000 человек;
- синдром Клайнфельтера (эндокринные нарушения, которые могут стать причиной мужского бесплодия) — 14–20 на 10 000;
- синдром Дауна — 9–13 на 10 000;
- синдром Тернера (болезнь, которая приводит к половому инфантилизму) — около 7 на 10 000;
- фенилкетонурия (нарушение метаболизма аминокислот) — до 3,8 на 10 000;
- нейрофиброматоз (заболевание, при котором у больного возникают опухоли) — около 3 на 10 000;
- муковисцидоз — 1–5 на 10 000;
- гемофилия — до 1,5 на 10 000[8].
Направления генетических обследований
Сегодня врачи выявляют генетические заболевания с высокой точностью, так как передовые технологии позволяют буквально заглянуть внутрь гена, определить, на каком уровне произошло нарушение.
На заметку
В зарубежной прессе уже появляются сообщения о том, что ведутся эксперименты по применению методов редактирования генома для борьбы с некоторыми заболеваниями. В частности, журнал Nature упоминал о подобных экспериментах в области борьбы с ВИЧ[9].
Есть несколько направлений обследований.
Диагностическое тестирование
Диагностическое тестирование проводится, если у пациента есть симптомы или особенности внешнего развития, служащие отличительной чертой генетического заболевания. Перед направлением на диагностическое тестирование проводят всесторонний осмотр пациента. Одна из отличительных черт наследственных заболеваний — это поражение нескольких органов и систем[10], поэтому при выделении целого ряда отклонений от нормы врач направляет пациента на молекулярно-генетическую диагностику.
Так как многие наследственные заболевания (например, синдромы Дауна, Эдвардса, Патау) связаны с нарушением количества хромосом (кариотипа), то для их подтверждения проводят кариотипирование, то есть изучение количества хромосом. Для анализа требуются клетки крови, которые в течение нескольких дней выращивают в особой среде, а затем окрашивают. Так врачи выделяют и идентифицируют каждую хромосому, определяют, нарушен ли их количественный состав[11], отмечают особенности внешнего строения.
Для выявления мутаций конкретных генов применяется метод ПЦР — полимеразной цепной реакции. Его суть состоит в выделении ДНК и многократном воспроизводстве интересующего исследователя участка. Как отмечают специалисты, преимущество ПЦР — его высокая точность: здесь почти невозможно получить ложноположительный результат. Метод удобен еще и тем, что для исследования может быть взята любая ткань организма[12].
Пренатальная и предимплантационная диагностика
Если вы знаете, что у вас в семье или в семье супруга были случаи наследственных болезней, то, конечно, захотите выяснить, какова вероятность проявления их у ваших детей. Врачи часто предлагают будущим родителям сделать пренатальную диагностику. А если пара использует вспомогательные репродуктивные технологии, то и предимплантационную генетическую диагностику плода (ПГД).
ПГД нужно сделать, если возраст матери превышает 35 лет, если у пары уже были прерывавшиеся беременности, а также родились дети с наследственными заболеваниями. Также врачи рекомендуют делать ПГД, если родители являются носителями генетического недуга. В этом случае в семье есть случаи проявления патологии, но сами супруги здоровы. А вот вероятность проявления болезни у ребенка может достигать 50%, причем ПГД помогает точно определить этот показатель. Анализ проводится, когда эмбрион, полученный «в пробирке», вырастает до стадии 6 или 8 клеток [13].
Пренатальная генетическая диагностика проводится, когда ребенок еще находится в утробе матери. Предположить наличие генетических отклонений врач может на основании анализов крови матери или по результатам УЗИ плода. Поэтому на начальном этапе беременная проходит трехмаркерный скрининг: в ее крови определяют уровень АФП, β-хорионического гонадотропина и эстриола. Если их концентрация отлична от нормы, то врач рекомендует выполнить генетическое обследование ребенка. Для этого с помощью пункции берут амниотическую жидкость и проводят кариотипирование плода. Единственный недостаток этого метода — долгий период ожидания результатов. Если последний будет негативным, то женщина просто может не успеть принять решение о прерывании беременности. Есть и альтернатива — анализ ворсин хориона. Его можно сделать на раннем сроке, но получение материала представляет угрозу для протекания беременности[14].
В последнее время появилась еще одна возможность пренатального обследования плода — неинвазивный пренатальный ДНК-тест (НИПТ-тест). В этом случае нужна только кровь матери. Точность теста достигает 99%, причем можно сделать обследование как на самые часто встречающиеся генетические патологии, так и полное исследование плода[15].
Определение носительства
Рассматривая виды наследования генетических заболеваний, мы упомянули об аутономно-рецессивном способе и о наследовании, сцепленном с полом. Человек может быть здоров, но в его генотипе при этом присутствует патологический ген. Выявить это помогает анализ на носительство. Многие делают его на стадии планирования беременности, чтобы вычислить вероятность рождения ребенка с генетическими заболеваниями.
Например, такая болезнь, как гемофилия, проявляется только у мужчин, женщины не болеют, но могут быть носителями. Поэтому женщинам, у которых есть родственники с проблемами свертывания крови, перед зачатием рекомендуется сделать скрининг гетерозиготного носительства, чтобы определить вероятность рождения мальчика с гемофилией[16].
Предсказательное генотипирование
И даже если у человека нет никаких признаков наследственных заболеваний, он все равно может пройти генетическую диагностику. Зачем? Дело в том, что только лишь нарушениями в генах определяются далеко не все наследственные заболевания. Ко многим патологиям может быть предрасположенность. Досимптоматическая диагностика, или ДНК-идентификация, выявляет ее[17]. Во многих клиниках это обследование носит название «генетический паспорт», его достаточно сделать один раз, потому что полученные результаты со временем не меняются.
По итогам ДНК-идентификации врач дает пациенту рекомендации: начиная от образа жизни и диеты и заканчивая профессиональными рисками. Следование им помогает избежать развития многих заболеваний.
Виды генетических заболеваний человека и ключевые методы их выявления
В зависимости от того, чем вызвано генетическое заболевание, врач выбирает и методы обследования пациента. Рассмотрим основные группы патологий.
Хромосомные болезни
Причиной этих генетических заболеваний служит нарушение в количественном составе хромосом или в их строении. Например, при наличии дополнительной (третьей) 21-й хромосомы формируется синдром Дауна. Причиной синдрома Шершевского-Тернера является наличие всего одной Х-хромосомы у женщин. А если у мужчины половые хромосомы присутствуют в сочетании XXY, а не XY, то ему ставится синдром Клайнфельтера.
Многие хромосомные нарушения, например, удвоение или утроение, несовместимы с жизнью. Чаще всего зародыши погибают в утробе, а родившиеся дети живут всего несколько дней[18]. В то же время бывают случаи, когда у человека есть разные виды клеток: несущие патологические хромосомы и не имеющие этих нарушений. Это явление носит название «мозаицизм», и тогда патология может проявляться в меньшей степени или практически не проявляться[19].
Для диагностики проводят кариотипирование. В качестве примера можно привести синдром Клайнфельтера — редкое генетическое заболевание, которым страдают мужчины. Внешне оно выражается в евнухоподобной внешности, увеличении грудных желез, нарушении половой функции. Подробное изучение состава половых хромосом помогает определить, какое именно нарушение произошло у пациента (лишних Х-хромосом может быть несколько). В зависимости от кариотипа варьируется и степень выраженности признаков заболевания [20].
Может быть нарушено и строение хромосом, а не только их количество. В процессе деления клеток, если «что-то пойдет не так», происходит утрата части хромосомы или, напротив, удвоение какого-либо участка. Хромосома может развернуться на 180 градусов (инверсия), или ее концы образуют кольцо. Например, синдром кошачьего крика — это следствие перестройки пятой хромосомы. Дети, родившиеся с такой патологией, специфически кричат (звук напоминает мяуканье кошки). Обычно они погибают в первые годы жизни, так как патология проявляется многочисленными пороками развития внутренних органов[21].
Пациентам с хромосомными заболеваниями назначают цитогенетическое обследование. Обычно ему подвергаются и родители, чтобы установить, имеет ли место наследуемая патология или же это единичный случай[22].
Генные мутации
Нарушения могут произойти не в хромосоме, а лишь на одном ее участке. Тогда мы говорим о генной мутации. Эти заболевания называются моногенными, к ним, в частности, относятся многие нарушения метаболизма: муковисцидоз, фенилкетонурия, андрогенитальный синдром и т.д. Многие из этих заболеваний могут быть выявлены при обязательном скрининге всех младенцев в роддоме. Ребенок, у которого есть отклонения от нормы, может быть направлен на дополнительное генетическое обследование. А принятые вовремя меры позволяют в некоторых случаях предотвратить развитие серьезных нарушений.
В то же время существуют заболевания, вызванные генными мутациями, которые не проявляются ярко и однозначно. В качестве примера можно привести синдром Вольфрама, который дебютирует как сахарный диабет в раннем возрасте, затем проявляется ухудшением зрения или слуха. Врач может подтвердить синдром только по результатам генетической экспертизы.
Мультифакториальные генетические болезни
Они выявляются при ДНК-идентификации. Анализ подтверждает наличие или отсутствие предрасположенности практически к любой патологии: от сахарного диабета до формирования различных зависимостей[23]. Так как роль генетических факторов и факторов внешней среды в развитии заболеваний различна не только для каждой патологии, но и для каждого пациента[24], рекомендации здесь могут быть только строго индивидуальными, сделанными на основании результатов анализов.
В последнее время нередки появления информации об экспресс-тестах, позволяющих определить нарушения в структуре ДНК непосредственно в день анализа. В частности, ученые из Дании создали «светящийся ДНК-тест», который дает результат в течение шести часов[25].
Где можно сдать анализы?
Наследственные заболевания отличаются большим разнообразием: это могут быть патологии, вызванные мутацией генов, нарушением строения хромосом, сочетанием нескольких факторов, в том числе факторов внешней среды. Именно поэтому генетическое обследование лучше выполнять в лаборатории, которая предоставляет максимально широкий спектр услуг. Желательно, чтобы в лаборатории проводилось и кариотипирование, и ПЦР, и пренатальная диагностика, и анализ на носительство.
Второй важный момент — наличие в лаборатории современного сертифицированного оборудования. Оно позволяет делать анализ максимально подробным и полным. Популярные экспресс-системы дают результат в тот же день, однако глубокий анализ генотипа им недоступен. Специализированные лаборатории предоставляют результаты через 2–3 дня, однако это более подробное и детализированное исследование, позволяющее точно установить и наличие заболевания, и предрасположенность к тем или иным патологиям.
Стоимость обследования в специализированной лаборатории во многом зависит от объема: при составлении генетического паспорта цена обследования может достигать 75 000–80 000 рублей[26].
Источник