
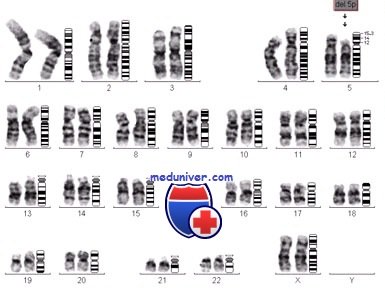
Кариотип больных с синдромом кошачьего крика

Синдром «кошачьего крика» – хромосомное нарушение, обусловленное делецией (отсутствием) фрагмента короткого плеча 5-ой хромосомы. Плач новорожденных с синдромом «кошачьего крика» по звуку напоминает кошачье мяуканье, что и послужило названию патологии. Кроме этого, у детей имеет место микроцефалия, лунообразное лицо, косоглазие, аномалии прикуса, различные врожденные пороки, грубое интеллектуальное недоразвитие и т. д. Синдром «кошачьего крика» диагностируется на основании совокупности характерных признаков и цитогенетического исследования. Специфического лечения синдрома «кошачьего крика» не существует; дети могут нуждаться в хирургической коррекции тяжелых врожденных аномалий.
Общие сведения
Синдром «кошачьего крика» (синдром Лежена) – частичная моносомия, связанная с нарушением структуры короткого плеча 5-ой хромосомы (потерей от 1/3 до 1/2 его длины, реже – полной утратой короткого плеча). Синдром «кошачьего крика» относится к числу редких хромосомных заболеваний с популяционной частотой 1:45-50 тыс. Среди новорожденных с синдромом «кошачьего крика» отмечается преобладание девочек над мальчиками в соотношении 4:3. Заболевание было описано в 1963 г. французским генетиком и педиатром Ж. Леженом и по автору получило название «синдром Лежена». Однако в литературе за данной патологией закрепилось образное название, связанное со специфическим признаком – плачем новорожденных, напоминающим кошачий крик.
Синдром кошачьего крика
Причины синдрома «кошачьего крика»
Развитие синдрома «кошачьего крика» связано с потерей фрагмента 5-ой хромосомы, а, следовательно, генетической информации, хранящейся на этом участке. В 85-90% случаев делеция короткого плеча образуется в результате случайной мутации, в 10-15% наследуется от родителей, являющихся носителями сбалансированной транслокации.
Наиболее частыми цитогенетическими вариантами хромосомной аберрации служат утрата одной трети или половины длины короткого плеча 5-ой хромосомы. Потеря меньшего участка или всего плеча встречается исключительно редко. При этом для степени выраженности клинической картины синдрома «кошачьего крика» важен не размер утерянного фрагмента, а отсутствие конкретного участка хромосомы. Так, при потере небольшого участка хромосомы в области 5p15.2 развиваются все клинические признаки синдрома, кроме кошачьего крика; критическим для возникновения характерного крика является выпадение участка хромосомы в области 5p15.3.
Наряду с простой делецией, могут встречаться другие цитогенетические вариации синдрома «кошачьего крика»: мозаицизм, кольцевая 5-я хромосома с делецией участка короткого плеча, реципрокная транслокация короткого плеча 5-ой хромосомы на другую хромосому.
Непосредственной причиной мутации могут выступать различные повреждающие факторы, воздействующие на половые клетки родителей либо на зиготу (алкоголь, курение, наркотические вещества, ионизирующая радиация, лекарственные препараты, химикаты и пр.). Вероятность появления ребенка с синдромом «кошачьего крика» выше в семьях, где уже рождались дети с подобным заболеванием.
Симптомы синдрома «кошачьего крика»
Новорожденные с синдромом «кошачьего крика», как правило, рождаются доношенными, но с небольшой пренатальной гипотрофией (средняя масса при рождении около 2500 г). Беременность у матери может протекать абсолютно нормально или сопровождаться угрозой самопроизвольного прерывания не чаще, чем в популяции. Наиболее патогномоничным ранним признаком синдрома является плач ребенка, который напоминает мяуканье кошки. Высокое и пронзительное звучание детского крика обусловлено анатомическими особенностями строения гортани при данном синдроме – узостью ее просвета, небольшим надгортанником, необычной складчатостью слизистой оболочки, мягкой консистенцией хрящей. Некоторые авторы считают, что специфический крик имеет центральное происхождение и не связан с недоразвитием гортани. Примерно у трети детей «кошачий крик» исчезает к 2-м годам, у остальных остается на всю жизнь.
Фенотип детей с синдромом «кошачьего крика» отличается преобладанием лицевой части черепа над мозговой, лунообразным лицом, гипертелоризмом, антимонголоидным разрезом глаз, эпикантом, деформацией ушных раковин, плоской спинкой носа, короткой шеей с крыловидными складками. При обследовании у детей выявляется микроцефалия, мышечная гипотония, снижение рефлексов, нарушение сосания и глотания. В неонатальном периоде может развиваться инспираторный стридор и цианоз.
Другие клинические проявления синдрома «кошачьего крика» могут значительно варьироваться по своему сочетанию у отдельных больных. Со стороны зрительной системы нередко обнаруживается врожденная катаракта, близорукость, косоглазие, атрофия зрительного нерва. Изменения со стороны костно-мышечной системы проявляются синдактилией стоп, врожденным вывихом бедра, косолапостью, плоскостопием, клинодактилией V пальца кисти, сколиозом, диастазом прямых мышц живота, паховыми и пупочными грыжами. Частыми спутниками синдрома «кошачьего крика» являются нарушения прикуса, «готическое» нёбо, микрогения, расщелины нёба и верхней губы, расщепление язычка.
У многих пациентов наблюдаются врожденные пороки сердца (ДМЖП, ДМПП, открытый артериальный поток, тетрада Фалло), пороки развития почек (гидронефроз, подковообразная почка), крипторхизм, гипоспадия. Реже отмечается мегаколон, запоры, кишечная непроходимость. Дерматоглифическими признаками синдрома «кошачьего крика» могут служить одна ладонная складка, поперечные складки сгибания и др.
Поведение детей характеризуется гиперактивностью, однообразными движениями, склонностью к агрессии и истерикам. Детям с синдромом «кошачьего крика» свойственна глубокая умственная отсталость в степени имбецильности и идиотии, грубое системное недоразвитие речи, выраженное отставание в моторном и физическом развитии.
Половая и репродуктивная функции у лиц с синдромом «кошачьего крика» обычно не страдают. Иногда у женщин выявляется двурогая матка, у мужчин – уменьшение размеров тестикул, однако сперматогенез существенно не нарушен, как, например, при синдроме Клайнфельтера.
Продолжительность жизни больных с синдромом «кошачьего крика» значительно укорочена; большая часть детей погибает в первый год жизни из-за сопутствующих пороков и их осложнений (чаще от сердечной и почечной недостаточности). Лишь около 10% доживают до подросткового возраста, хотя имеются отдельные сообщения о больных, достигших 50 лет.
Диагностика синдрома «кошачьего крика»
Если в семье уже имелись случаи хромосомных заболеваний, еще на этапе планирования беременности будущим родителям рекомендуется посетить генетика и пройти генетическое тестирование. В процессе беременности наличие у плода синдрома «кошачьего крика» может быть заподозрено на основании результатов ультразвукового пренатального скрининга. В этом случае для окончательного подтверждения хромосомной аномалии рекомендуется проведение инвазивной пренатальной диагностики (амниоцентеза, биопсии ворсин хориона или кордоцентеза) и непосредственного анализа генетического материала плода.
После рождения предварительный диагноз синдрома «кошачьего крика» устанавливается неонатологом на основании типичных диагностических признаков (характерного плача, фенотипических черт, множественных стигм дизэмбриогенеза). Для подтверждения хромосомной патологии проводится цитогенетическое исследование.
Учитывая наличие у детей с синдромом «кошачьего крика» множественных аномалий развития, необходимо, чтобы в первые дни жизни новорожденные были осмотрены детским кардиологом, детским офтальмологом, детским урологом, детским ортопедом и другими специалистами.
Лечение синдрома «кошачьего крика»
Специфического лечения данного хромосомного заболевания в настоящее время не существует. Для стимуляции психомоторного развития под наблюдением детского невролога проводятся курсы медикаментозной терапии, массажа, физиотерапии, ЛФК. Дети с синдромом «кошачьего крика» нуждаются в помощи психологов, дефектологов, логопедов.
Врожденные пороки сердца при синдроме «кошачьего крика» часто требуют хирургической коррекции, поэтому детям необходима консультация кардиохирурга, проведение ЭхоКГ и других необходимых исследований. Дети с патологией мочевыделительной системы должны находиться под наблюдением детского нефролога и периодически проходить комплекс необходимых обследований (УЗИ почек, общий анализ мочи, биохимическое исследование крови и мочи и др.).
Прогноз и профилактика синдрома «кошачьего крика»
На продолжительность и качество жизни больных влияет тяжесть самого синдрома и сопутствующих врожденных пороков, уровень оказания медицинской и психолого-педагогической помощи. В целом долговременный прогноз неблагоприятный. При специальном обучении дети имеют словарный запас, достаточный для бытового общения, однако по уровню психомоторного развития обычно не поднимаются выше дошкольников.
Профилактика синдрома «кошачьего крика» заключается в тщательной подготовке к беременности и исключении возможных неблагоприятных воздействий на организм родителей еще задолго до зачатия. При рождении в семье ребенка с синдромом «кошачьего крика», родители в обязательном порядке должны пройти цитогенетическое обследование для исключения носительства реципрокной сбалансированной транслокации.
Источник
Синдром кошачьего крика: признаки, фенотипУ пациентов с дисморфиями описано множество цитогенетически обнаруживаемых делеций, но эти делеций часто бывают только у нескольких пациентов и не связаны с общепризнанными синдромами. Тем не менее есть множество хорошо описанных синдромов делеций аутосом, в которых серия пациентов имеет ту же или аналогичную делецию, заканчивающуюся ясно различимым синдромом. В целом цитогенетически видимые аутосомные делеций происходят с предполагаемой встречаемостью 1 на 7000 живых новорожденных.
Один из таких синдромов — синдром «кошачьего крика», терминальная или интерстициальная делеция части короткого плеча хромосомы 5. Делециидали такое название из-за того, что плач новорожденного с этим синдромом похож на мяуканье кошки. Синдром составляет приблизительно 1% всех стоящих на учете умственно отсталых больных. Характерные лицевые особенности показаны на рисунке, сочетаются с микроцефалией, гипертелоризмом, эпикантом, низко посаженными ушными раковинами, иногда с преаурикулярными придатками и микрогнатией. Другие симптомы — умственная отсталость от умеренной до тяжелой степени и пороки сердца.
Большинство случаев синдрома «кошачьего крика» спорадические; 10-15% пациентов — потомки носителей транслокации. Точки разрыва и протяженность утраченного сегмента хромосомы 5р у разных пациентов различны, но критической областью, делетированной у всех пациентов с клинической симптоматикой, является участок 5р15. При использовании FISH и CGH-матричного анализа хромосом (см. главы 4 и 5) обнаружено множество генов, утрачиваемых при del(5p), и начала проясняться основа взаимоотношений моносомии этих генов с фенотипом. Большинство клинических признаков оказалось следствием гаплонедостаточности генов в пределах участка 5р15.2, а аномалия, вызывающая синдром «кошачьего крика» вызвана делецией гена или генов в пределах небольшого участка 5р15.3. Степень умственной отсталости обычно согласуется с размером делеций, хотя анализ CGH-матриц указывает, что несоразмерно выраженную умственную задержку может вызывать гаплонедостаточность конкретных регионов в участке 5р14-р15. Фенотипическая карта иллюстрирует, как геномные методы могут уточнить общую концепцию соотношения генотипа и фенотипа в клинической цитогенетике, что становится важным как для понимания патофизиологии изменений, так и для генетического консультирования при многих повторяющихся хромосомных аномалиях. – Также рекомендуем “Геномные болезни. Особенности микроделеционных и дупликационных синдромов” Оглавление темы “Хромосомные патологии”:
|

Источник
Синдром кошачьего крика (МКБ-10: Q93.4, синдром Лежена) — это наследственное заболевание, обусловленное утратой части пятой хромосомы. Младенцы с этим заболеванием часто имеют пронзительный крик, напоминающий кошачий, небольшой размер головы и характерный внешний вид лица. Люди с этим заболеванием могут отставать в физическом и психическом развитии.
Наиболее опасными проявлениями синдрома кошачьего крика являются дефекты развития сердца: дефекты межжелудочковой или межпредсердной перегородок, тетрада Фалло и открытый артериальный проток. От тяжести дефектов сердца и уровня оказываемой медицинской помощи зависит продолжительность и качество жизни больного. Диагноз ставится на основании клинических симптомов и подтверждается результатами генетического тестирования.
Лечение является симптоматическим. Может понадобиться хирургическое исправление пороков сердца.
Синдром обусловлен делецией короткого плеча пятой хромосомы. В этом районе расположены гены CTNND2 и TERT, а также некоторые другие.
Синонимы: Синдром Лежена, синдром делеции короткого плеча хромосомы 5, cat cry syndrome, chromosome 5p deletion syndrome, cri-du-chat syndrome.
Синдром кошачьего крика впервые описал врач Ж. Лежен (J. Lejeune) с соавторами в 1963 году у трех детей с множественными аномалиями, умственной отсталостью и характерным плачем, который напоминал кошачий крик [1].
Распространенность и тип наследования
Распространенность синдрома кошачьего крика в мире составляет 1 случай на 45 000 человек [2]. Частота встречаемости данного заболевания в России не отличается от общемировой.
Этот синдром встречается гораздо чаще других синдромов, связанных с делециями аутосом. Около 85 % всех случаев заболевания являются спорадическими, 15 % наследуются от фенотипически нормальных родителей — носителей сбалансированных перестроек [3].
Чаще всего синдром кошачьего крика диагностируется при рождении ребёнка. Врачей неонатологов может насторожить характерный плач ребёнка, напоминающий кошачье мяуканье. Причиной такого плача является изменение (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки) или недоразвитие гортани [4]. Признак чаще всего исчезает к концу первого года жизни [5]. Также могут наблюдаться такие симптомы, как низкая масса тела при рождении и мышечная гипотония, лунообразное лицо с широко расставленными глазами. Глазные щели расположены под небольшим углом — от переносицы вбок и вниз, может наблюдаться косоглазие.
По мере роста ребёнка наблюдается физическое и психическое отставание в развитии [6].
Дефекты развития сердца могут включать в себя: дефект межпредсердной или межжелудочковой перегородки, открытый артериальный проток, тетраду Фалло [4]. Дефекты могут быть диагностированы с помощью ЭКГ и ЭхоКГ.
Во всех случаях при подозрении на синдром кошачьего крика необходимо обратиться к врачу-генетику. Больным и их родителям показано цитогенетическое обследование. Без своеобразного крика надежный диагноз до цитогенетического исследования установить невозможно, так как большинство клинических симптомов этой болезни встречаются и при других хромосомных аномалиях.
Необходимо постоянное наблюдение со стороны педиатра и психоневролога. Врачами рекомендуются средства, стимулирующие психомоторное развитие ребенка, гимнастика и лечебный массаж [3].
Также необходимы консультации хирурга и кардиолога для оценки возможности исправления пороков развития сердца.
Профилактика заключается в своевременном проведении генетического консультирования семей, где имелись больные с синдромом кошачьего крика. Необходимо определить кариотипы родителей больного ребёнка. Наличие изменений короткого плеча 5-й пары хромосом является абсолютным показанием для определения кариотипа плода при последующих беременностях путём амниоцентеза и исследования амниотических клеток. Сбалансированная транслокация у одного из родителей требует также исследования кариотипа у его кровных родственников с целью выявления лиц, имеющих транслокацию.
Синдром кошачьего крика объясняется частичной моносомией пятой хромосомы, он развивается при делеции (с утратой от трети до половины, реже при полной утрате) короткого плеча хромосомы [7]. Для развития клинической картины синдрома имеет значение не величина утраченного участка, а потеря конкретного короткого фрагмента хромосомы. Изредка отмечается мозаицизм по делеции или образование кольцевой хромосомы 5 [8].
Одним из генов, которые находятся в дистальной части короткого плеча 5-й хромосомы (локус 5p15.33), является ген TERT. Он кодирует фермент теломеразную обратную транскриптазу, которая важна для поддержания длины теломер и устойчивой пролиферации клеток. Делеция TERT специфически вовлечена в развитие клинической картины синдрома кошачьего крика [9]. Лимфоциты пациентов с этим синдромом имеют более короткие теломеры, чем у здоровых людей соответствующего возраста. В настоящее время ученые предполагают, что неспособность поддерживать длину теломер может быть одним из генетических факторов, вносящих вклад в развитие синдрома кошачьего крика [9].
Другой ген, расположенный в дистальной части короткого плеча 5-й хромосомы (локус 5p15.2), — ген CTNND2. Он кодирует дельта-катенин — белок, экспрессируемый на ранней стадии развития головного мозга. CTNND2 участвует в контроле подвижности клеток. Утрата этого гена ведет к развитию умственной недостаточности при синдроме кошачьего крика [11].
- CTNND2 (5p15.2): делеция гена
- TERT (5p15.33): делеция гена
- Lejeune, J., Lafourcade, J., Berger, R., Vialatta, J., Boeswillwald, M., Seringe, P., Turpin, R. Trois ca de deletion partielle du bras court d’un chromosome 5. C. R. Hebd. Seances Acad. Sci. 257: 3098, 1963. PubMed: 14095841
- Niebuhr, E. The cri du chat syndrome: epidemiology, cytogenetics, and clinical features. Hum. Genet. 44: 227-275, 1978. PubMed: 365706
- Nguyen, J. M., Qualmann, K. J., Okashah, R., Reilly, A., Alexeyev, M. F., Campbell, D. J. 5p deletions: current knowledge and future directions. Am. J. Med. Genet. 169C: 224-238, 2015. PubMed: 26235846
- Cerruti Mainardi, P., Perfumo, C., Cali, A., Coucourde, G., Pastore, G., Cavani, S., Zara, F., Overhauser, J., Pierluigi, M., Dagna Bricarelli, F. Clinical and molecular characterisation of 80 patients with 5p deletion: genotype-phenotype correlation. J. Med. Genet. 38: 151-158, 2001.
- Van Buggenhout, G. J. C. M., Pijkels, E., Holvoet, M., Schaap, C., Hamel, B. C. J., Fryns, J. P. Cri du chat syndrome: changing phenotype in older patients. Am. J. Med. Genet. 90: 203-215, 2000. PubMed: 10678657
- Kjaer, I., Niebuhr, E. Studies of the cranial base in 23 patients with cri-du-chat syndrome suggest a cranial developmental field involved in the condition. Am. J. Med. Genet. 82: 6-14, 1999. PubMed: 9916835
- Overhauser, J., Huang, X., Gersh, M., Wilson, W., McMahon, J., Bengtsson, U., Rojas, K., Meyer, M., Wasmuth, J. J. Molecular and phenotypic mapping of the short arm of chromosome 5: sublocalization of the critical region for the cri-du-chat syndrome. Hum. Molec. Genet. 3: 247-252, 1994. PubMed: 8004090
- Zhang, X., Snijders, A., Segraves, R., Zhang, X., Niebuhr, A., Albertson, D., Yang, H., Gray, J., Niebuhr, E., Bolund, L., Pinkel, D. High-resolution mapping of genotype-phenotype relationships in cri du chat syndrome using array comparative genomic hybridization. Am. J. Hum. Genet. 76: 312-326, 2005. PubMed: 15635506
- Zhang, A., Zheng, C., Hou, M., Lindvall, C., Li, K.-J., Erlandsson, F., Bjorkholm, M., Gruber, A., Blennow, E., Xu, D. Deletion of the telomerase reverse transcriptase gene and haploinsufficiency of telomere maintenance in cri du chat syndrome. Am. J. Hum. Genet. 72: 940-948, 2003. PubMed: 12629597
- South, S. T., Swensen, J. J., Maxwell, T., Rope, A., Brothman, A. R., Chen, Z. A new genomic mechanism leading to cri-du-chat syndrome. Am. J. Med. Genet. 140A: 2714-2720, 2006. PubMed: 17103439
- Medina, M., Marinescu, R. C., Overhauser, J., Kosik, K. S. Hemizygosity of delta-catenin (CTNND2) is associated with severe mental retardation in cri-du-chat syndrome. Genomics 63: 157-164, 2000. PubMed: 10673328
Источник