
Клиника и генетика синдрома марфана

Синдром Марфана: причины, диагностика, лечениеЭтиология и встречаемость синдрома Марфана. Синдром Марфана (MIM №154700) — панэтническое аутосомно-доминантное заболевание соединительной ткани, вызванное мутациями в гене фибриллина 1 (FBN1, MIM № 134797). Синдром Марфана имеет встречаемость около 1 на 10 000. Приблизительно 25-35% пациентов имеют новую мутацию. Мутации, вызывающие синдром Марфана, разбросаны по гену, и каждая мутация обычно уникальна в семье. Патогенез синдрома МарфанаГен FBN1 кодирует фибриллин 1, внеклеточный матричный гликопротеид с широким распределением. Фибриллин 1 полимеризуется, формируя микрофибриллы как в эластичных, так и в неэластичных тканях, например стенке аорты, цилиарных поясках и коже. Мутации влияют на синтез, процессинг, секрецию, полимеризацию или устойчивость фибриллина 1. Исследования накопления и экспрессии фибриллина 1 в культуре клеток предположили доминантный отрицательный патогенез, т.е. синтез мутантного фибриллина 1 тормозит образование нормальных микрофибрилл нормальным фибриллином 1 или стимулирует протеолиз несоответствующих внеклеточных микрофибрилл. Последние исследования на мышиных моделях синдрома Марфана указывают, что половинного количества нормального фибриллина 1 недостаточно, чтобы проводить эффективную сборку микрофибрилл. Таким образом, патогенезу болезни также может содействовать гаплонедостаточность. Кроме синдрома Марфана, мутации в гене FBN1 могут вызывать другие синдромы, включая неонатальный синдром Марфана, изолированные скелетные симптомы, аутосомно-доминантную эктопию хрусталиков и фенотип MASS (марфаноидные симптомы, включая пролапс митрального клапана или миопию, пограничное и непрогресирующее расширение аорты, и неспецифические изменения скелета и кожи). В общем, фенотипы довольно схожи в пределах семьи, хотя тяжесть фенотипических проявлений может значительно изменяться. До настоящего времени точное соотношение между генотипом и фенотипом не определено. Внутрисемейная и межсемейная изменчивость позволяет предполагать, что в определении фенотипа важную роль играют окружающая среда и эпигенетические факторы. Последние исследования на мышиных моделях показывают, что фибриллин 1 не просто структурный белок, и что синдром Марфана не просто результат структурной слабости тканей. Более того, микрофибриллы фибриллина 1 в норме связывают и уменьшают концентрацию и активность факторов роста суперсемейства TGFb. Потеря фибриллина 1 увеличивает сигналы свободного TGFb значительно содействующие заболеванию, так как антагонисты TGFb устраняют легочные и клапанные изменения, наблюдаемые у мышей с недостаточностью фибриллина 1.
Фенотип и развитие синдрома МарфанаСиндром Марфана — мультисистемное заболевание со скелетными, глазными, сердечно-сосудистыми, легочными, кожными и другими аномалиями. Скелетные аномалии включают очень высокий рост (отношение размаха рук к росту >1,05; соотношение верхнего и нижнего сегментов <0,85 у взрослых), арахнодактилию, аномалии грудины, сколиоз, разболтанность суставов, готическое нёбо. Аномалии глаз включают подвывих хрусталиков, уплощение роговицы, удлинение глазного яблока и гипоплазию радужки. Сердечнососудистые аномалии включают пролапс митрального клапана, аортальную регургитацию и расширение и расслаивающую аневризму восходящей аорты. Легочные аномалии включают спонтанный пневмоторакс и расширение концевых пузырьков. Аномалии кожи включают атрофические бороздки и рецидивирующие грыжи. Аномалии твердой мозговой оболочки включают выбухание оболочки в крестцово-поясничном отделе. Большинство признаков синдрома Марфана появляются с возрастом. Скелетные аномалии типа аномалии грудины и сколиоза ухудшаются с ростом костей. Подвывих хрусталика часто присутствует уже в раннем детстве, но может развиваться и в юности. С повышенной частотой при синдроме Марфана встречаются отслойка сетчатки, глаукома и катаракты. Сердечно-сосудистые осложнения обнаруживаются в любом возрасте и развиваются в течение всей жизни. Основные причины преждевременной смерти пациентов с синдромом Марфана — сердечная недостаточность вследствие регургитации клапанов и аневризмы и разрыва аорты. Тем не менее в связи с улучшением хирургической и терапевтической помощи при аневризме аорты выживание улучшилось. С 1972 по 1993 г. ожидаемый возраст выживания для 50% пациентов поднялся с 49 до 74 лет для женщин и с 41 до 70 лет для мужчин. Особенности фенотипических проявлений синдрома Марфана:
Лечение синдрома МарфанаСиндром Марфана — клинический диагноз, определяемый по наличию конкретных симптомов. Подтверждение синдрома Марфана идентификацией мутаций в гене FBN1 в настоящее время практически нецелесообразно, поскольку крайняя аллельная гетерогенность делает идентификацию причинно-обусловленной мутации в каждой семье крайне трудозатратной, а также из-за недостаточно надежной корреляции между генотипом и фенотипом. Анализ мутаций имеет недостаточную чувствительность или специфичность для синдрома Марфана, что ограничивает его клиническую пользу. Для синдрома Марфана недоступно эффективное лечение; следовательно, помощь сфокусирована на профилактике осложнений и симптоматическом лечении. Оказание офтальмологической помощи включает регулярные осмотры, коррекцию миопии и, часто, замену хрусталика. Ортопедическая помощь заключается в укрепляющем лечении или хирургической коррекции сколиоза. Помощь при аномалии грудины в основном косметическая. Физиотерапия может компенсировать нестабильность суставов. Сердечно-сосудистые проблемы решаются комбинацией терапевтических и хирургических мероприятий. Терапевтические усилия направлены на предохранение или замедление развития расширения корня аорты за счет уменьшения кардиологических показателей, снижения артериального давления и усилия выброса желудочков с помощью бета-адреноблокаторов, ограничение участия в контактных видах спорта, соревновательных видах спорта и в изометрических упражнениях. Профилактическая замена корня аорты показана, когда расширение аорты или аортальная регургитация становится достаточно тяжелой. Большинству пациентов в настоящее время проводят надклапанную замену корня аорты, не требующую постоянного приема противосвертывающих препаратов. Гемодинамические изменения, связанные с беременностью, могут приводить к прогрессирующему расширению или расслоению аорты. Полагают, что расслоение аорты вторично к гормональным изменениям, увеличению объема крови и сердечного выброса, связанных с беременностью и родами. Современные исследования считают, что риск беременности слишком велик, если ширина корня аорты превышает 4 см. Женщины могут выбрать проведение надклапанной замены аорты перед беременностью. Риски наследования синдрома МарфанаПациенты с синдромом Марфана имеют 50% риск иметь ребенка, больного синдромом Марфана. В семьях, передающих синдром Марфана, членов семьи, находящихся в группе риска, можно выявлять, либо обнаруживая мутацию (в тех редких случаях, когда она известна), либо анализом сцепления, если маркеры, тесно сцепленные с локусом FBN1, имеют очевидную связь с болезнью в семье пробанда. Пренатальная диагностика доступна только для семей, в которых возможны исследования сцепления или известна мутация в гене FBN1. Пример синдрома Марфана. Д.Л., здоровый 16-летний ученик средней школы, звезда баскетбола, направлен в генетическую клинику для обследования по поводу синдрома Марфана. Телосложение Д.Л. похоже на телосложение его отца. Отец Д.Л., высокий субтильный человек, умер во время утренней пробежки; у других членов семьи случаев скелетных аномалий, внезапной смерти, снижения зрения или врожденных аномалий не было. При медицинском осмотре выявлены астеническое телосложение, высокое дугообразное нёбо, небольшая деформация грудины по типу «куриной» груди, арахнодактилия, соотношение размах рук/рост 1,1, диастолический шум и стрии на плечах и бедрах. Эхокардиография выявила расширение корня аорты с аортальной регургитацией. Офтальмологическое обследование показало двусторонний иридодонез и легкое смещение хрусталиков кверху. На основе медицинского осмотра и результатов обследования генетик объяснил пациенту и его матери, что у него синдром Марфана. – Также рекомендуем “Синдром Миллера-Дикера: причины, диагностика, лечение” Оглавление темы “Наследственные болезни”:
|
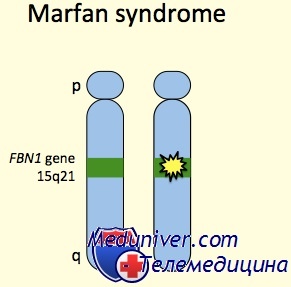
Источник

Синдром Марфана — генетически детерминированное заболевание, характеризующееся поражением или недоразвитием соединительнотканных волокон во время эмбриогенеза и проявляющееся дисфункциональными изменениями со стороны зрительного анализатора, костно-суставной и кардиоваскулярной систем.
Обычно синдром передается по наследству от родителей детям по аутосомно-доминантному принципу. В некоторых случаях он становится результатом мутации гена, кодирующего синтез фибриллина и оказывающего влияние на формирование одновременно нескольких фенотипических признаков. Генетические изменения – мутации происходят под воздействием негативных эндогенных и экзогенных факторов. Белок фибриллин является важной составной частью многих структур организма. При его недостатке соединительная ткань теряет свою прочность и эластичность, что отражается на состоянии сосудистой стенки и связочно-суставного аппарата.
Синдром был открыт в 1875 году офтальмологом из Америки Э. Вильямсом. Он обнаружил полное смещение хрусталика глаза от своего нормального положения у брата и сестры, которые имели высокий рост и чрезмерную подвижность суставов с рождения. Спустя несколько лет детский врач из Франции А. Марфан вел наблюдения за девочкой 5 лет с прогрессирующими аномалиями скелета, чрезмерно длинными конечностями и «паучьими пальцами». Он дал четкое описание патологии, благодаря чему синдром получил его имя.

мальчик с синдромом Марфана
Клиническая симптоматика синдрома весьма полиморфна. Это связано с наличием пораженной соединительной ткани во всех внутренних структурах организма. Системная соединительнотканная недостаточность проявляется признаками поражения скелета, сердца и сосудов, глаз, кожи, ЦНС, легких. Подобные патологические изменения формируются внутриутробно у плода. Симптоматика синдрома варьируется от стертых форм до процессов, несовместимых с жизнью.
Больные имеют диспропорциональные конечности, рост выше среднего, вытянутые пальцы, гипермобильные суставы, худощавое тело, готическое небо, неправильный прикус, глубоко расположенные глаза. Они страдают гигантизмом, миопией, эктопией хрусталика, изменением формы грудины, кифосколиозом. Клинические признаки синдрома обусловлены гиперрастяжимостью тканей.

Синдром Марфана — наследственная коллагенопатия, встречающаяся у 1 из 10000-20000 человек. Патология обнаруживается повсеместно, среди людей любой национальности, пола и места проживания – одинаково часто как среди жителей юга, так и севера. В семейных парах, где отцу больше 35 лет, чаще рождаются больные дети.
Диагностика патологии основывается на данных наследственного анамнеза, визуального осмотра, результатах дополнительных исследований. Пациенты с синдромом Марфана внешне похожи друг на друга. Специалисты по внешнему виду могут предположить наличие недуга. Лечение болезни медикаментозное и оперативное, заключающееся в устранении структурных и функциональных нарушений в сердечной мышце, зрительном анализаторе, костно-суставном аппарате. Оперативное вмешательство показано в тех случаях, когда лекарственное и физиотерапевтическое лечение не дает положительных результатов. Если недуг не лечить, продолжительность жизни больных ограничится 30—40 годами. Причиной их смерти становится разрыв аорты или острая коронарная недостаточность. Современная медицина позволяет пациентам успешно лечиться и полноценно жить до самой старости.
Этиологические факторы
Синдром Марфана – врожденная аномалия, отличающаяся следующими генетическими признаками:
-
 аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;
аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении; - выраженным плейотропизмом — множественным действием гена;
- варьирующей экспрессивностью – степенью развития признака, контролируемого данным геном;
- высокой пенетрантностью – вероятностью фенотипического проявления признака при наличии соответствующего гена.
Синдром развивается в результате мутации гена, кодирующего биосинтез особого белка фибриллина — важной структуры межклеточного вещества, которая обеспечивает эластичность и сократительную способность соединительнотканных волокон. Мутация происходит спонтанно в момент зачатия в яйцеклетке или сперматозоиде. При недостатке фибриллина нарушается процесс формирования волокон. Он перестают быть прочными и упругими, становятся чрезмерно растяжимыми и менее устойчивыми к деформациям. В наибольшей степени повреждению подвержены сосуды и связки.
Фибриллин необходим для работы цинновой связки, с помощью которой хрусталик прикрепляется к ресничному телу. При дефиците белка эта связка ослабляется, что проявляется миопией, подвывихом хрусталика, вторичной глаукомой, снижением остроты зрения. Кроме зрительного анализатора, белок фибриллин содержится в связках аорты и обеспечивает ее устойчивость к нагрузкам. При ослаблении этих связок происходит расширение сосуда и расслоение его стенок. Подобные изменения являются смертельно опасными для человека. При синдроме Марфана часто отмечается поражение двустворчатого клапана, что требует проведения хирургической коррекции.
Классификация
Формы патологии:
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
Этиологическая классификация:
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
Симптоматика
Синдром Марфана проявляется разнообразными клиническими признаками, что связано с присутствием соединительнотканных волокон в различных структурах организма. У больных появляются признаки поражения костей и суставов, зрительного анализатора, сердца, сосудов, нервов, легких, кожного покрова.
Симптомы дисфункциональных расстройств внутренних органов возникают на фоне сильной усталости и быстрой утомляемости после незначительных физических нагрузок, миалгии, вялости, мышечной гипотонии, приступообразной цефалгии. По мере роста и развития организма человека черты заболевания становятся более выраженными.

Скелет
Клинические признаки синдрома Марфана, обусловленные поражением костей, мышц и связок:
- короткое туловище и длинные ноги,
- молоткообразные пальцы стопы,
- паучьи пальцы кисти,
- худощавое телосложение,
- гипотонус мышц,
- удлиненное и узковатое лицо,
- глубокая посадка глаз,
- отсутствие зазоров между зубами,
- большой нос,
- микрогнатия,
- недоразвитые скулы,
- большие и низко расположенные уши,
- «готическое» небо,
- прогения,
- гиперподвижность суставов,
- деформированная грудная клетка в виде воронки или киля птицы,
- искривление позвоночника,
- смещение вышележащего позвонка относительно нижележащего,
- уплощение сводов стопы,
- продавливание вертлужной впадины.

Сердце и сосуды
Поражение сердечной мышцы при синдроме Марфана определяет его исход. Обычно возникают дефекты в структуре аорты, легочного ствола, развиваются пороки развития клапанов сердца:
- дилатация предсердий и желудочков,
- мешкообразное расширение ограниченного участка аортальной стенки,

- пролабирование двустворчатого клапана,
- миксоматоз сердца,
- кардиомиопатия,
- разрыв хорд митрального клапана,
- кальциноз аортального клапана,
- различные виды аритмии – мерцательная, экстрасистолия,
- воспаление внутренней оболочки сердца – эндокарда,
- кардиалгия с иррадиацией в спину, ключицу, руку, плечо,
- похолодание конечностей,
- затрудненное дыхание,
- сердечные шумы,
- на ЭКГ — признаки ИБС.
Глаза
У лиц с синдромом Марфана развивается патология зрения:
- миопия,
- смещение хрусталика в сторону,
- уплощение роговой оболочки глаза и ее утолщение,
- недоразвитие радужки и ресничной мышцы,
-
 страбизм,
страбизм, - катаракта,
- асимметричность зрачков,
- глаукома,
- астигматизм,
- дальнозоркость,
- колобома глаза,
- спазмирование сосудов сетчатки и высокий риск ее отслойки.
Прочие органы
Поражение нервной системы:
- ишемические и геморрагические инсульты,
- субарахноидальное кровотечение,
- выпячивание оболочек спинного мозга.
Ослабление и растяжение дурального мешка проявляется болью в пояснице, ногах, тазу и другими неврологическими признаками, цефалгией.
Поражение бронхопульмональной системы:
- скопление воздуха между висцеральным и париетальным листками плевры,
- эмфизема легких,
- удлинение и перерастяжение альвеол,
- ночное апноэ,
- рак легких.

У больных пневмотораксом воздух скапливается в плевральной полости, а легкие сжимаются. Их дыхание становится частым и поверхностным, в груди болит, кожные покровы сначала бледнеют, а затем приобретают синюшный оттенок.
Поражение кожи, мягких тканей, внутренних органов:
- смещение почки в каудальном направлении,
- цистоцеле,
- пролапс матки,
- кисты и новообразования в печени и почках,
- варикоз,
- стрии на коже,
- грыжи.
Больные с синдромом Марфана легко возбудимы, гиперактивны, чрезмерно эмоциональны, часто плаксивы и разражительны. Они имеют неординарные способности и высокий уровень интеллекта.
«Готическое» небо и микрогнатия приводят к возникновению речевых нарушений. При поражении скелета и суставов появляются артралгии и миалгии, развивается ранний остеоартрит.
Синдром Марфана проявляется по-разному у лиц, обращающихся за медицинской помощью. У одних выявляются ярко выраженные признаки патологии, у других симптоматика бывает стертой. Прогрессирование синдрома происходит по мере взросления человека. Клинические проявления определяются локализацией и интенсивностью поражения органов и систем.
Методы диагностики

проявления синдрома Марфана в младшем возрасте
Выявлением синдрома Марфана занимаются специалисты в области генетики, кардиологии, офтальмологии, неврологии, ортопедии. Диагностика патологии включает сбор анамнеза жизни и болезни, выявление типичных клинических признаков, анализ внешнего осмотра и физикальных данных, результатов кардиографического и рентгенографического обследования, посещения офтальмолога, составление родословной у генетика.
Основные диагностические методики:
- общий анализ крови и мочи — типичные признаки воспаления;
- биохимическое исследование крови позволяет выявить дисфункцию определенного органа, возникшую в результате развития патологического процесса, а также определить первопричину заболевания и назначить правильное лечение;
- электрокардиография отображает электрические потенциалы, сформированные в работающем сердце;
- эхокардиография – исследование морфологических и функциональных изменений сердца и его клапанного аппарата;
- рентгенографическое и томографическое исследование — информативные диагностические методики, обнаруживающие поражение костей, суставов, внутренних органов и мягких тканей;
- аортография – рентгенологического исследования аорты с использованием контрастного вещества;
- УЗИ внутренних органов,
- биомикроскопия и офтальмоскопия,
- молекулярно-генетический анализ.
Методы лечения

Синдром Марфана неизлечим. Этиотропной терапии недуга не существует, ведь невозможно заменить гены ребенка. Больным проводят симптоматическую терапию, целью которой является облегчение общего состояния, устранение симптомов и предупреждение тяжелых осложнений.
Медикаментозное лечение:
- β-адреноблокаторы – «Пропранолол», «Атенолол»;
- блокаторы кальциевых каналов – «Нифедипин», «Верапамил»;
- ингибиторы АПФ – «Каптоприл», «Лизиноприл»;
-
 коллагеннормализующая терапия – «Алфлутоп», «Румалон», «Структум»;
коллагеннормализующая терапия – «Алфлутоп», «Румалон», «Структум»; - метаболики – «Рибоксин», «Милдронат»;
- поливитаминные комплексы;
- антибиотики для профилактики инфекционного эндокардита;
- антикоагулянты для профилактики тромбоза;
- антиоксиданты и антигипоксанты – «Коэнзим Q10», «Элькар»;
- ноотропные препараты – «Пирацетам», «Винпоцетин».
При снижении остроты зрения пациентам с офтальмологическими заболеваниями прописывают очки для постоянного ношения или контактные линзы.
Хирургическое лечение:
- оперативное вмешательство на сердце и аорте — протезирование аорты и клапанов сердца, реконструктивные и пластические операции,
- операции на глазах — коррекция близорукости лазером, замена хрусталика, устранение глаукомы,
- хирургическая коррекция скелета — пластика грудной клетки при ее воронкообразной деформации, стабилизация позвоночника, протезирование крупных суставов.
Людям с марфаноидным фенотипом стертой формы показано физиолечение и ЛФК.
Клинические рекомендации специалистов своим пациентам:
- ведение здорового образа жизни,
- ограничение тяжелого труда и физического перенапряжения,
- отказ от спортивных игр повышенной активности,
- регулярное посещение специалистов в области кардиологии, офтальмологии, ортопедии, неврологии,
- нахождение больных под постоянным наблюдением и контролем врачей,
- периодическое прохождение диагностического обследования.
Прогноз и профилактические мероприятия

Прогноз синдрома Марфана неоднозначный. Он зависит от состояния кардиоваскулярной системы, глаз и скелета больного. При наличии серьезных нарушений со стороны этих органов длительность жизни пациентов ограничивается 40-50 годами, и повышается риск внезапной смерти. С помощью своевременной кардиохирургической коррекции можно улучшить качество жизни больных и вернуть им трудоспособность.
Все пары, в семейном анамнезе которых имелись случаи наследственных заболеваний, должны перед беременностью посетить генетика и заранее сдать все необходимы анализы. Пренатальная диагностика — комплекс мероприятий, проводимых с целью выявления патологии на стадии внутриутробного развития. Она заключается в проведении УЗИ плода и биохимического скрининга материнских сывороточных маркеров. К ее инвазивным методам относятся: биопсия ворсин хориона, исследование околоплодных вод, пуповинной крови и клеток плаценты.
Всем больным необходимо вовремя санировать имеющиеся очаги хронической инфекции в организме — лечить кариес, тонзиллит, синусит с помощью антибиотиков. Это крайне важное мероприятие, поскольку у лиц с синдромом Марфана иммунитет ослаблен. Им следует закаляться, правильно питаться, оптимизировать режим дня, полноценно спать, подолгу гулять на свежем воздухе, бороться с вредными привычками, избегать конфликтных ситуаций и стрессов.
Видео: синдром Марфана в программе “Жить здорово!”
Видео: ток-шоу о синдроме Марфана
Источник