
Код мкб синдром секкеля

Синдром Секкеля является унаследованной формой врожденной карликовости , а это значит, что младенец начинает очень мало и не может нормально расти после рождения. В то время как люди с синдромом Секкеля обычно будут пропорциональны по масштабу, у них будет отчетливо малый размер головы. Также распространена умственная отсталость.

Несмотря на множество физических и умственных проблем, с которыми сталкивается человек с синдромом Секкеля, многие из них, как известно, хорошо живут более 50 лет.
Причины синдрома Секкеля
Синдром Секкеля является наследственным расстройством, связанным с генетическими мутациями на одной из трех разных хромосом. Считается, что это бывает крайне редко, с немногим более 100 случаев, зарегистрированных с 1960 года. Многие дети, получившие диагноз синдрома Секкеля, родились у родителей, которые тесно связаны (родственники), например, брак с кузенами или братьями и сестрами.
Синдром Секкеля является рецессивным генетическим заболеванием, он возникает только тогда, когда ребенок наследует один и тот же ненормальный ген от каждого родителя. Если ребенок получает один нормальный ген и один ненормальный ген, ребенок будет носителем синдрома, но обычно не проявляет симптомов.
Если оба родителя имеют одинаковую хромосомную мутацию для синдрома Секкеля, их риск иметь ребенка с синдромом Секкеля составляет 25%, а риск наличия носителя — 50%.
Характеристики синдрома Секкеля
Синдром Секкеля характеризуется аномально медленным развитием плода и низким весом при рождении.
После рождения у ребенка будет медленный рост и созревание костей, что приводит к короткому, но пропорциональному росту (в отличие от ахондроплазии). Лица с синдромом Секкеля обладают различными физическими и характеристиками развития, в том числе:
Очень маленький размер и вес при рождении
- Чрезвычайно малый, пропорциональный рост
- Аномально небольшой размер головы (микроцефалия)
- Клювообразный выступ носа
- Узкое лицо
- Нарушенные уши
- Необычно маленькая челюсть (микрогнатия)
- Умственная отсталость, часто тяжелая с IQ менее 50
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, повреждение зубов и другие деформации кости. Расстройства крови, такие как анемия (низкие эритроциты), панцитопения (недостаточно клеток крови) или острый миелоидный лейкоз.
В некоторых случаях семенники у мужчин не спускаются в мошонку, а женщины могут иметь аномально увеличенный клитор. Кроме того, у людей с синдромом Секкеля могут быть чрезмерные волосы на теле и единая глубокая складка на ладонях (известная как складки обезьян).
Диагностика синдрома Секкеля
Диагноз синдрома Секкеля основан почти исключительно на физических симптомах. Возможно, потребуется рентгеновское изображение и другие инструменты (МРТ, КТ), чтобы отличить его от других подобных условий. В настоящее время нет лабораторных или генетических тестов, специфичных для синдрома Секкеля. В некоторых случаях окончательный диагноз не может быть сделан до тех пор, пока ребенок не станет старше и не появятся характерные симптомы.
Лечение синдрома Секкеля
Лечение синдрома Секкеля сосредоточено на любой медицинской проблеме, которая может возникнуть, особенно при нарушениях крови и структурных деформациях. У людей с психическими расстройствами и их семьям должна быть предоставлена соответствующая социальная поддержка и консультационные услуги.
Источник
Что такое синдром Секкеля?
Синдром Секкеля чрезвычайно редкое наследственное заболевание, характеризующееся задержкой роста до рождения (задержка внутриутробного развития), что приводит к низкой массе тела при рождении. Задержка роста продолжается после рождения (послеродовой период), что приводит к низкорослости (карликовость). Другие симптомы и физические особенности, связанные с синдромом Секкеля, включают аномально маленькую голову (микроцефалия); разную степень умственной отсталости; и/или необычные характерные черты лица, включая «клювоподобный» выступ носа. Другие черты лица могут включать аномально большие глаза, узкое лицо, деформированные уши и/или необычно маленькую челюсть (микрогнатия). Кроме того, у некоторых младенцев может наблюдаться постоянная фиксация пятых пальцев в согнутом положении (клинодактилия), деформация (дисплазия) тазобедренного сустава, вывих кости в предплечье (радиальный смещение) и/или другие физические отклонения.
Признаки и симптомы
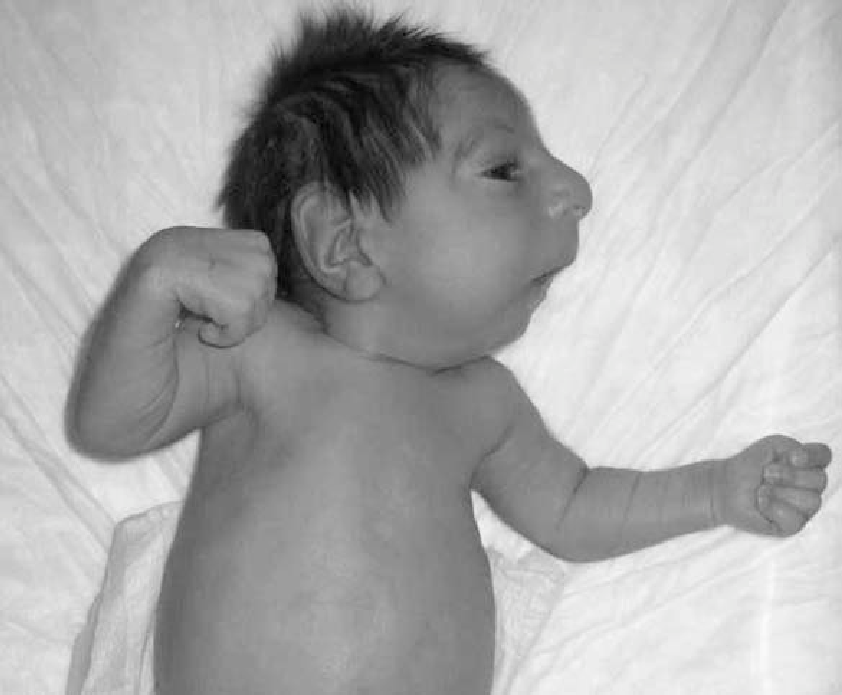
Синдром Секкеля характеризуется аномально медленным развитием плода и низкой массой тела при рождении. После рождения у ребенка будет наблюдаться медленный рост и созревание костей, что приведет к низкому, но пропорциональному росту (в отличие от карликовости коротких конечностей или ахондроплазии). Больные с синдромом Секкеля имеют различные физические отклонения и особенности развития, в том числе:
- Очень маленький размер и вес при рождении (в среднем 3,3 фунта или 1,4 кг);
- Чрезвычайно маленький, пропорциональный рост;
- Аномально маленький размер головы (микроцефалия);
- Клювовидный выступ носа;
- Узкое лицо;
- Деформированные уши;
- Необычно маленькая челюсть (микрогнатия);
- Умственная отсталость, часто тяжелая, с IQ (коэффициент интеллекта) менее 50.
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, пороки развития зубов и другие деформации костей. Также часто наблюдаются такие заболевания крови, как анемия (низкий уровень эритроцитов), панцитопения (недостаток клеток крови) или острый миелоидный лейкоз (тип рака крови).
В некоторых случаях у мужчин яички не могут опускаться в мошонку (крипторхизм), а у женщин может быть ненормально увеличенный клитор. Кроме того, у больных с синдромом может быть чрезмерное оволосение на теле и одна единственная глубокая складка на ладонях (известная как обезьянья складка).
Причины
Синдром Секкеля — чрезвычайно редкое заболевание, передающееся по аутосомно-рецессивному типу. Три варианта синдрома Секкеля включают нарушения или изменения (мутации) генов на трех разных хромосомах. Расположение генной карты: синдром Секеля 1 на хромосоме 3 (3q22-q24); Синдром Секкеля 2 на хромосоме 18 (18p11.31-q11) и синдром Секеля 3 на хромосоме 14 (14q21-q22).
Хромосомы, присутствующие в ядре клеток человека, несут генетическую информацию для каждого человека. Клетки человеческого тела обычно имеют 46 хромосом. Пары человеческих хромосом пронумерованы от 1 до 22, а половые хромосомы обозначены X и Y. У мужчин одна X и одна Y хромосома, а у женщин две X хромосомы. У каждой хромосомы есть короткое плечо, обозначенное буквой «p», и длинное плечо, обозначенное буквой «q». Хромосомы далее подразделяются на множество пронумерованных полос. Например, «хромосома 3q22-q24» относится к области между полосой 22 и полосой 24 на длинном плече хромосомы 3. Подобным образом «хромосома 18p11.31-q11.2» относится к более широкой области между полосой 11p31 на коротком плече хромосомы 18 и полосе 11.2 на длинном плече хромосомы 18. Опять же, хромосома 14q21-q22 относится к более узкой области на длинном отрезке хромосомы 14 между полосами 21 и 22. Пронумерованные полосы указывают расположение тысяч генов, присутствующих на каждой хромосоме.
Специфический ген, участвующий в синдроме Секкеля 1, известен как ген атаксии-телеангиэктазии и ген Rad3-родственного белка (ATR). Гены, участвующие в синдроме Секкеля 2 и 3 типов, неизвестны.
Генетические заболевания определяются комбинацией генов определенного признака, которые находятся на хромосомах, полученных от отца и матери.
Рецессивные генетические расстройства возникают, когда человек наследует один и тот же аномальный ген одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, он будет носителем болезни, но обычно бессимптомным. Риск для двух родителей-носителей передать дефектный ген и, следовательно, иметь больного ребенка, составляет 25% при каждой беременности. Риск иметь ребенка, который будет носителем, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей и будет генетически нормальным по данному признаку, составляет 25%. Риск одинаков для мужчин и женщин.
Все люди несут несколько аномальных генов. Родители, которые являются близкими родственниками (кровными родственниками), имеют больше шансов, чем неродственные родители, иметь один и тот же аномальный ген, что увеличивает риск рождения детей с рецессивным генетическим заболеванием.
Затронутые группы населения
Синдром Секкеля — чрезвычайно редкое наследственное заболевание, которое поражает в равной степени мужчин и женщин. Точная частота расстройства неизвестна. С момента первоначального описания в 1960 г. в медицинской литературе было зарегистрировано более 100 случаев.
Близкие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Секкеля. Сравнения могут быть полезны для дифференциальной диагностики:
Синдром Секкеля — один из шести расстройств в классе карликовости, известном как «изначальная карликовость». Эти расстройства имеют схожие характеристики, включая пороки развития скелета (дисплазию), дефицит роста до рождения (задержка внутриутробного развития) и в младенчестве и детстве, что в конечном итоге приводит к разной степени низкого роста. В эту группу заболеваний в настоящее время входят пять расстройств: синдром Секкеля, синдром Мейера-Горлина; Синдром Рассела-Сильвера; Остеодиспластическая примордиальная карликовость Маевского I/III; и остеодиспластическая примордиальная карликовость Маевского II типа.
Остеодиспластическая примордиальная карликовость Маевского II типа является чрезвычайно редким генетическим заболеванием, характеризующимся низким ростом, низкой массой тела при рождении, аномально маленькой головой (микроцефалия) и/или аномалиями скелета. Другие физические признаки могут включать большие глаза, выступающий нос в форме клюва, опущенную челюсть и/или узкое лицо. Обычно возникает серьезная недостаточность роста до рождения (внутриутробная недостаточность роста). В некоторых случаях может присутствовать умственная отсталость. Также могут появиться различные дополнительные симптомы. Конкретные симптомы и степень тяжести варьируются от случая к случаю. Считается, что II тип остеодиспластической примордиальной карликовости Маевского наследуется по аутосомно-рецессивному типу.
Диагностика
С появлением технически совершенной ультасонографии синдром Секкеля можно диагностировать до рождения (пренатально). При ультразвуковом исследовании плода используются отраженные звуковых волны для создания изображения развивающегося плода. После рождения можно заподозрить синдром Секкеля на основании тщательного клинического обследования, подробного анамнеза пациента и различных специализированных тестов. Хотя характерные черепно-лицевые, скелетные и/или другие физические аномалии, связанные с синдромом Секкеля, могут присутствовать при рождении, в некоторых случаях диагноз может не подтверждаться до тех пор, пока больной ребенок не вырастет и не разовьется полный синдром (например, когда станет очевидным низкий рост и/или умственная отсталость).
Низкий рост, связанный с синдромом Секкеля, предполагает пропорциональный рост рук и ног, что позволяет проводить дифференциальную диагностику от синдромов, связанных с низким ростом и аномально маленькими руками и ногами (карликовость с короткими конечностями).
Стандартные методы лечения
От синдрома Секкеля нет лекарства. Некоторые лекарства могут быть назначены для устранения других симптомов, связанных с заболеванием.
Лечение направлено на решение любой медицинской проблемы, которая может возникнуть, особенно заболеваний крови и структурных деформаций. Людям с умственными недостатками и их семьям потребуется соответствующая социальная поддержка и консультационные услуги.
Прогноз
Дети, страдающие синдромом Секкеля, могут жить в течение длительного периода времени, хотя они часто сталкиваются с глубокими психическими и физическими отклонениями.
Источник
Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Секкеля – лечение в Италии
Синдром Секкеля (синонимы: карликовость с «птицеголовостью», примордиальная карликовость с микроцефалией) – редкое наследственное заболевание, характеризующееся пре- и постнатальным отставанием в росте, микроцефалией с задержкой психического развития и характерным изменением лица, напоминающим «птицеголовость».
Заболевание, по всей вероятности, было известно давно, так как термины «птицеголовая карликовость» и «наноцефалия» принадлежат Н.Зеске в 1960 г. опубликовал статью, в которой привел результаты обследования двух собственных пациентов и литературные данные о 24 больных, имевших сходные клинические проявления
Синдром Секкеля – редкий синдром, характеризующийся тяжелой низкорослостью, низким весом при рождении, очень маленькой головой (микроцефалия), большим лбом, большими глазами, низкими ушами, большим носом и небольшим подбородком. Дефекты костей рук и ног, вывихи локтевых суставов и бедер, неспособность выпрямлять колени и крипторхизм, также часто встречаются у детей с этим синдромом.
Хромосомная нестабильность и недопроизводство всех типов клеток крови (панцитопения) отмечаются у некоторых пациентов. Это заболевание является генетическим. Оно наследуется по аутосомно-рецессивному типу. Синдром Секкеля является неоднородным генетическим заболеванием, которое может быть связано с мутациями в генах, расположенных на хромосомах 3 и 18.
Следует отличать синдрос Секкеля от другиз заболеваний с низкорослостью. Для этого проводится дифференциальная диагностика при характерных дополнительных симптомах.
Низкая масса тела при рождении в случае доношенной беременности, внутриутробная задержка роста. Этот частый симптом свидетельствует о наличии генетически обусловленного снижения потенции роста тканей, недостаточности плаценты или же нарушений роста во внутриутробном периоде токсического или воспалительного генеза. Если отсутствует дополнительная симптоматика и вместе с тем имеющуюся клиническую картину нельзя отнести к какому-либо определенному заболеванию, говорят о примордиальной низкорослости. При отсутствии других симптомов последняя описывается как семейное заболевание.
В спорадических случаях следует думать о необратимом внутриутробном повреждении редупликационного потенциала клеток неизвестным патогенным фактором. При чисто примордиальной низкорослости дифференцировка скелета соответствует хронологическому возрасту.
- а) Примордиальная низкорослость: синдром Рассела – Сильвера.
- б) Синдром Секкеля: гетерогенная группа примордиальных карликов с микроцефалией, большим носом, скошенным подбородком и интеллектуальным недоразвитием.
- в) Прогерия (синдром Гатчинсона-Джилфорда): замедление роста и развитие начиная со 2-3-го года жизни, преждевременное старение, атрофия кожи, выпадение волос.
- г) Синдром Блума: телеангиэктатическая эритема щек, долихоцефалия.
- д) Синдром Корнелии де Ланге; умственное недоразвитие, густые, сросшиеся над переносицей брови, микроцефалия, усиленное оволосение тела.
- е) Лепрехаунизм (синдром Донохуа): отсутствие или недоразвитие подкожной жировой клетчатки, гипертрофия клитора или полового члена, усиленное оволосение.
- ж) Синдром Фанкони: панцитопения, гипоплазия I пальца руки или нарушение его закладки, пигментные аномалии.
- з) Семейная, или полигенная, низкорослость, при которой в большинстве случаев наблюдается только легкая задержка роста во внутриутробном периоде.
- и) Трисомия 18.
Очень короткие конечности (увеличенное отношение верхнего сегмента к нижнему). К этой группе прежде всего относятся заболевания скелета, обусловленные поражением эпифизов и метафизов. При дифференциальной диагностике обязательно следует подумать об атиреозе или тяжелом гипотиреозе (особенно учитывая возможности их лечения). Фосфатный диабет также может сопровождаться легкой диспропорциональной низкорослостью с преобладанием верхнего сегмента. Пропорции могут существенно меняться в процессе роста. Здесь представлены сведения о пациентах старшего детского и юношеского возраста в соответствии с обобщением Спрангера.
- а) Ахондроплазия (хондродистрофия): относительно большая голова с провалившимся основанием носа, особенное укорочение предплечий и голеней, кифоз, типичные рентгенологические находки.
- б) Гипохондроплазия: слабо выраженная диспропорциональная низкорослость, переразгибание суставов, люмбальный лордоз, типичные рентгенологические данные.
- в) Псевдоахондроплазия: пропорции, как и при ахондроплазии, но с нормальным формированием мозгового и лицевого черепа, относительно позднее по сравнению с ахондроплазией проявление заболевания, отсутствие типичных рентгенологических симптомов со стороны таза.
- г) Синдром Эллиса-Ван-Кревельда: порок сердца, дистрофия ногтей и пальцев стоп, эписпадия.
- д) Синдром Маркезани: изменения хрусталиков, как при синдроме Марфана (дрожание хрусталиков, может быть шаровидный хрусталик), брахидактилия, возможны легкие контрактуры.
- е) Метафизарная хондродисплазия, тип Шмидта: укорочение трубчатых костей (больше проксимальных), грибовидно вздутые эпифизы с размытыми очертаниями.
- ж) Метафизарная хондродисплазия, тип Мак-Кусика: редкие тонкие волосы, брови и ресницы, переразгибание суставов, типичная рентгенологическая картина.
- з) Метафизарная хондродисплазия, тип Енсена: утолщенные суставы с костным ограничением подвижности, формирование косолапости стоп, типичная рентгенологическая картина.
- и) Дисхондростеоз (синдром Лери – Вейля): укорочение предплечья и лучевой кости по отношению к локтевой, дорсальный, поддающийся редрессации подвывих дистального конца локтевой кости (деформация Маделунга).
- к) Пикнодизостоз: короткие концевые фаланги, большой мозговой череп, большие роднички (которые могут быть открытыми до зрелого возраста), спонтанные переломы.
- л) Карликовость с микромелией: гипоплазированные локтевая и малоберцовая кости, а также нижняя челюсть.
- м) Карликовость с микромелией: характерная “треугольная” деформация большеберцовой кости.
- н) Псевдогипопаратиреоз: ожирение, обращающее на себя внимание круглое лицо, укороченные пальцы рук, изменения метаболизма.
- о) Псевдопсевдогипопаратиреоз.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку – так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 –
срочное лечение в Италии
ЗАПРОС в КЛИНИКУ
Источник