
Микроделеционные синдромы что это такое

Несколько десятков лет назад, любой микроделеционный синдром воспринимался как патология неизвестного происхождения и не рассматривался как хромосомное отклонение. Дело в том, что у ученых не было возможности провести тонкую и точную диагностику наследственных заболеваний, не навредив здоровью матери и не повлияв на процесс развития плода.
Благо сегодня, для установления причины «сбоя» не требуется проводить забор плодных тканей или околоплодных вод. Ведь генетикам удалось установить, что с 8 недели беременности, в крови беременной женщины появляются фрагменты фетальной ДНК, способные дать огромный массив данных о возможных отклонениях в развитии будущего ребенка!
Синдром микроделеции — что это?
В отличие от моно- или трисомии, микроделеции приводят к уничтожению мелких фрагментов хромосом, провоцируя различные генетические аномалии. К списку самых распространенных заболеваний, подпадающих под группу микроделеционных синдромов, можно отнести:
- Делецию хромосомы 1p36, характеризующуюся пороками развития, умственными отсталостями, задержкой роста, внезапными судорогами, нарушениями работы органов чувств, дисморфизм черепно-лицевой области;
- Болезнь Вольфа-Хиршхорна, провоцирующую развитие сердечной и почечной недостаточности, задержкой развития, микроцефалией, нарушениями черт лица и т.д.;
- Синдром кошачьего крика, характеризующий отставанием в развитии, низкой массой при рождении, выраженной мышечной гипотонией. Основным признаком является характерный плач, напоминающий мяуканье кошки в результате изменения гортани;
- Болезнь Сотоса, выраженная высокорослостью, ускоренным ростом (до 4-5 лет), нарушением координации движения, тремором и судорожными приступами. Пациенты подвержены развитию онкологии.
В общей сложности, существует более 20 заболеваний, характеризующихся отсутствием отдельных фрагментов хромосом.
Основные причины возникновения
По результатам исследований, из 25 детей, родившихся с признаками, свойственными делеции, каждый 2-й ребенок унаследует болезнь от родителей. Из остальных, 40% зафиксированных случаев не имеют связи с наследственностью, в то время как у оставшихся 10% установить причины проявления болезней невозможно. Более того, некоторые заболевания (например – синдром Ди Джорджи) диагностируются в довольно позднем возрасте, из-за чего человек может жить с заболеванием, даже не догадываясь об этом.
Последствия хромосомного отклонения
К сожалению, большинство неинвазивных пренатальных тестов ДНК нацелены на выявление трисомии 13, 18, 21 хромосом и неспособны выявить микроделеционные аномалии. А ведь своевременная диагностика позволяет установить причину появления болезни и разработать эффективный способ её решения: от коррекции рациона питания, до работы над социализацией больного, страдающего от задержки развития.
В отсутствие своевременной диагностики и адекватного лечения, начальные симптомы могут развиться в более тяжелую форму, снижая не только качество, но и продолжительность жизни людей, страдающих от подобных недугов.
С недавних пор, жители нашей страны получили возможность пройти анализ ДНК на предмет отсутствия генетических мутаций, передающихся по наследству. Ведь в распоряжении специалистов лаборатории INLABgenetics имеется передовое материально-техническое оснащение, позволяющее выделять необходимые маркеры безболезненным и абсолютно безопасным способом: образцом для дородового генетического скрининга служит венозная кровь матери.
Для получения дополнительной информации об услуге достаточно позвонить по номеру «горячей линии» или оставить запрос на e-mail: менеджеры нашей компании готовы ответить на любой интересующий вас вопрос!
Источник
Группа хромосомных патологий, называемых «синдромы микроделеции 22q11.2», – одна из причин возникновения умственной отсталости у детей. Такие нарушения не всегда удается зафиксировать методами цитогенетической диагностики, но можно выявить с помощью ультразвукового исследования.

Сергей Адольфович, что представляют собой микроделеционные синдромы?
Микроделеционные синдромы (МДС) – это особый вид хромосомных заболеваний, при которых происходит потеря микроскопического участка хромосомного материала. Стоит особо остановиться на микроделеции участка q11.2 хромосомы 22, которая является одной из самых распространенных у человека. Задача выявления данной группы синдромов достаточно сложна из-за различных форм проявления заболевания. Клинически синдром может проявляться судорогами, возможны затруднения при вскармливании, новорожденные имеют повышенный риск внезапной смерти. Наиболее же частыми причинами смерти детей с этим синдромом в раннем возрасте являются тяжелые врожденные пороки сердца и инфекции, а выжившие дети часто отстают в физическом и умственном развитии.
Какова частота возникновения таких патологий?
Эта аномалия встречается с относительно высокой частотой – 1 случай на 4000–7000 живорожденных. Это второй по частоте синдром после синдрома Дауна. Но если для последнего фактором риска рождения больного ребенка является возраст матери, то МДС 22q11.2 не зависит от возраста женщины. И совсем молодые женщины (до 30 лет) могут быть подвержены риску рождения детей с подобными отклонениями, при этом очень большое количество случаев остается не диагностированным ни во время беременности, ни даже после рождения ребенка.
Чем опасны микроделеционные синдромы?
Подобные хромосомные нарушения могут развиваться с минимальными проявлениями. К примеру, если во время скринингового исследования обнаруживается изолированный порок сердца (кстати, такие патологии диагностируются с помощью УЗИ уже в первом триместре – со срока 11 недель и 2 дня), беременную женщину в дальнейшем, как правило, направляют к специалисту-кардиологу для определения хирургической тактики. Другие же проявления микроделеционных синдромов, которые поражают не только сердечно-сосудистую, но и дыхательную, иммунную системы и влияют на умственное развитие ребенка, могут остаться незамеченными. Причем некоторые симптомы начинают проявляться в возрасте около 5 лет, когда выясняется, что ребенок, которого нужно готовить к школе, необучаемый и страдает умственной отсталостью. А между тем, чем позже ставится точный диагноз, тем тяжелее последствия МДС для ребенка, так как могут формироваться физические отклонения, опасные для жизни, задержка развития и проблемы в школе.

Как этого избежать?
Врач-диагност, выявляя определенные пороки развития сердца плода, обязательно должен рассматривать все признаки и прежде всего обратить внимание на развитие тимуса (вилочковой железы). Здесь важны не только знания, но и большой опыт вместе с мануальными навыками и качеством ультразвукового оборудования. Возможности, предоставляемые клиникой, позволяют мне совершенствоваться в этом направлении.
Есть ли другие маркеры МДС?
Опасность микроделеций состоит еще и в том, что только 75–80% развивающихся синдромов сопровождаются ярко выраженными пороками сердца. Остальные имеют менее заметные проявления, которые также обязательно нужно отслеживать и учитывать уже в первом триместре, и тут важную роль играют лицевые аномалии. В условиях нашей клиники мне удалось получить патент на изобретение способа ранней диагностики лицевых аномалий и связанных с ними синдромов.
Что нужно делать будущей матери во время беременности, чтобы родить здорового ребенка?
Для начала необходимо исключить все возможные осложнения. Нужно провести подробную и качественную диагностику первого триместра, оценить принадлежность к группе риска, особенно если предыдущие беременности окончились неудачно, так как более 1/4 случаев заболевания являются семейными. МДС 22q11.2 не является исключительной редкостью, и долю больных с этим синдромом можно оценить как 5–10% среди всех детей, нуждающихся в оперативном лечении врожденного порока сердца, и 5% среди детей, имеющих расщелину нёба, не сочетающуюся с расщелиной губы. Огромную роль играет грамотная консультация семьи генетиком с использованием новейших знаний и возможностей лабораторной диагностики.
Дата публикации: 12.09.16
Источник
.jpg)
Микроделеционные синдромы – довольно большое число заболеваний, которые воздействуют на самые разные компоненты организма. Причина возникновения данных заболеваний кроется в потерях (делециях) крошечных частей хромосом. Часто такие отклонения сложно диагностировать при сдаче стандартных анализов. Однако сегодня, с развитием науки, появляется все большее количество возможностей диагностики этих синдромов.
Диагностика
Преимплантационная генетическая диагностика (ПГД) – метод диагностики заболеваний, который проводится до беременности и позволяет выявить генетические мутации. Возможен только при проведении ЭКО. После проведения процедуры на нескольких эмбрионах, отбираются наиболее здоровые, которые рекомендуются для переноса в матку.
Неинвазивное пренатальное тестирование (НИПТ) – анализ, который проводится после 10 недель беременности. Для его проведения достаточно лишь венозной крови, однако он способен выявить практически все патологии в развитии хромосом ребенка.
Лаборатория Медикал Геномикс рекомендует расширенный неинвазивный пренатальный тест VERAGENE
Получить бесплатную консультацию
Ответим на все вопросы по неинвазивному пренатальному тесту ДНК
Описание самых распространенных микроделеционных синдромов
Синдром делеции хромосомы 1p36
Это врожденное заболевание, встречающееся примерно у 1 младенца из 5000. Данная патология вызвана потерей короткого плеча хромосомы 1. Основные характеристик заболевания: задержки в умственном развитии ребенка разной степени, отклонения в физическом развитии и росте. Также характерны следующие особенности внешности: глубокая посадка глаз, плоский нос, ассиметричное лицо, небольшие уши и рот. У таких детей часто возникают проблемы с речью, вплоть до полного ее отсутствия. Обычно они агрессивны по отношению к другим людям, вспыльчивы, проявляют признаки аутизма. Проявление перечисленных симптомов зависит от степени повреждения хромосомы.
Синдром Вольфа – Хиршхорна
Эта болезнь проявляется у 1 из 50 000 младенцев и является генетической. Обычно люди с ней долго не живут. В среднем, около 30 лет, однако при особо тяжелых формах ребенок может не прожить и года. Причиной служит потеря короткого плеча 4 хромосомы, тяжесть случая зависит от размера повреждения. У большинства больных патология проявляется «de novo», примерно 90% случаев. У 10% это вызвано транслокацией у родителей. Для больных характерна задержка всех форм развития: умственное, физическое, психологического. Часто проявляются серьезные заболевания внутренних органов: сердца и почек. Обычно дети рождаются с очень маленьким весом: при нормальном протекании беременности он достигает 2 кг. Обладают специфической внешностью: деформация губ, ушных раковин, нос необычной формы, напоминающей клюв.
Синдром кошачьего крика
Проявляется болезнь после утраты большей части короткого плеча 5-й хромосомы. С ним рождается 1 из 20 000 детей. Проявление симптомов синдрома влияет на продолжительность жизни ребенка. Характеризуется отставаниями в умственном и физическом развитии. Для внешности характерны широко посаженные глаза, необычная форма ушей и разрез глаз, складки у глаз и ушей. После рождения плач ребенка напоминает кошачье мяуканье, из-за чего синдром и получил свое название. Вызвано это деформированием гортани, однако проходит после года жизни. Обычно дети рождаются очень маленького веса и часто страдают от врожденных пороков внутренних органов.
Синдром Смита – Магениса
С этим заболеванием рождается 1 из 25 000 детей. Характеризуется особенностями в развитии черепа и строения тела: широкий лоб и переносица, очень близко расположенные брови, рот необычной формы, аномалии развития ушей. Распространены такие заболевания, как сколиоз, порок сердца, почечная недостаточность, проблемы с развитием головного мозга. У детей проявляется умственная отсталость, трудность с речью, а средний уровень IQ равен 40- 55. Наблюдаются отклонения в поведении: дети часто пытаются навредить себе. Также сон больных очень чуткий и прерывистый. Это часто является причиной вспышек гнева.
Синдром Ди Джорджи
Проявляется у 1 из 4000 детей. Возникает эта мутация de novo. Сопровождается снижением иммунитета, проблемами с сосудами и сердцем. Таким детям могут требоваться операции для поддержания сердечного здоровья, регулярные визиты к кардиологам и постоянный прием необходимых препаратов. У больных людей присутствуют отличительные черты лица: небольшой рот, слегка увеличена переносица. Однако они могут выражены очень слабо, или не выражены вообще. Из-за ослабленного иммунитета дети с этим синдромом больше других подвержены таким заболеваниям, как ангина или пневмония. Однако с возрастом иммунная система организма может прийти в норму.
Синдром Ангельмана
С ним рождается 1 из 10 000 младенцев. При этом заболевании в организме нет ряда генов 15 – й хромосомы. Стоит отметить, что при возникновении синдрома Ангельмана страдает именно материнская хромосома. Обычно у этих детей есть проблемы с набором веса и питанием, начиная с грудничкового возраста. У них также часто наблюдается задержка в физическом развитии: проблемы с ходьбой. Практически у каждого ребенка проблемы с устным выражением мыслей: они могут все понимать, но не уметь правильно сказать. Характеризуются также гиперактивностью, проблемами с обучаемостью, склонностью к эпилепсии. Они также обладают специфической внешностью, которую называли «лицом марионетки»: сравнительно небольшой размер головы, крупный рот и немного выдвинутый вперед подбородок, большой промежуток между зубами и иногда высунутый наружу язык.
Синдром Прадера – Вилли
При этом синдроме наблюдается такое же нарушение 15-й хромосомы, как и при синдроме Ангельмана с одним существенным отличием: в данном случае страдает отцовская хромосома. Во время беременности характерно неправильное расположение ребенка, невысокий уровень подвижности. У детей в раннем возрасте наблюдается склонность к лишнему весу. Страдает координация, возможно затруднение физического развития: медленный рост, небольшие конечности. Также люди с этой патологией скорее всего страдают сколиозом, у них плохое качество костей и зубов, обычно они бесплодны. Могут проявляться задержки в речи и психическом развитии. Однако все эти признаки вряд ли будут у одного больного, обычно в одном случае проявляется не более 5.
Источник
[16-018]
Исследование микроделеций и микродупликаций хромосом
5400 руб.
Микродупликационные/микроделеционные синдромы (ММС) представляют собой гетерогенную группу генетических нозологий, характеризующихся изменением количества копий участка хромосом. Считается, что ММС являются одной из главных причин синдромального и несидромального спорадического отставания в развитии.
Синонимы русские
Микроделеции и микродупликации.
Синонимы английские
Microdeletion and microduplication.
Метод исследования
Полимеразная цепная реакция, фрагментный анализ, MLPA.
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Микродупликационные/микроделеционные синдромы (ММС) представляют собой гетерогенную группу генетических нозологий, характеризующихся изменением количества копий участка хромосом. Синдромы данной группы чаще всего характеризуются нарушением когнитивных функций, задержкой речевого развития и задержкой роста, различного рода стигмами, дисморфизмами и мальформациями, затрагивающими широкий спектр систем и органов. Считается, что ММС являются одной из главных причин синдромального и несидромального спорадического отставания в развитии.
Исследование на основные формы микроделеционных и микродупликационных синдромов позволяет комплексно и одновременно выявлять 26 синдромов, такие как синдром ДиДжорджи, синдром Прадера – Вилли, синдром Ангельмана, синдром кошачьего крика, 1р36 делеция и другие. Рекомендуется проводить данное исследование в качестве первичного скрининга у пациентов с синдромальными и несиндромальными формами отставания в развитии.
Данный тест детектирует мутации, характерные для следующих нозологий: 1р36 микроделеционный синдром, 2p16.1-p15 микроделеционный синдром, 2q23.1 микроделеционный/микродупликационный синдром, 3q29 микроделеционный/микродупликационный синдром, 9q22.3 микроделеционный синдром, LIS1-ассоциированная лиссэнцефалия (синдром Миллера – Дикера / изолированная лиссэнцефалия / синдром двойной коры), SATB2 – ассоциированный синдром, нейрофиброматоз 1-го типа, KANSL1-связанная умственная отсталость, синдром Виттевеена – Колька, синдром Вольфа – Хиршхорна, синдром ДиДжорджи / велокардиофациальный синдром, синдром дупликации 15q, синдром дупликации гена MECP2, синдром кошачьего крика, синдром Лангера – Гидеона (трихоринофалангеальный синдром 2-го типа), синдром Прадера – Вилли / синдром Ангельмана, синдром Рубинштейна – Тейби, синдром Смита – Магениса, синдром Сотоса, синдром Фелана – МакДермида, синдром Вильямса – Бойрена, синдром Потоцки – Лупски, синдром Клайнфельтера, синдром Шерешевского – Тернера, синдром тройной Х хромосомы.
Для чего используется исследование?
- Диагностика микроделеционных и микродупликационных заболеваний
Когда назначается исследование?
- При подтверждении причин отставания и задержки развития.
- При дифференциальной диагностике причин пороков развития сердца, печени, почек, нервной системы.
- При подтверждении диагноза “микроделеционный” или “микродупликационный синдром”.
Что означают результаты?
Наличие патогенной микроделеции или микродупликации подтверждает диагноз “ММС”.
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды и индивидуальных генетических факторов.
Важные замечания
- Для получения заключения по результату исследования необходимо проконсультироваться у клинического генетика.
Кто назначает исследование?
Педиатр, невролог, ортопед.
Источник

Клинические проявления микроделеционных синдромов

Синдромы с микроделециями Синдром Локализация микроделеции Делеция 1 р36 1 p 36 Вильямса 7 q 11 Лангера-Гидеона 8 q 34 WAGR (опухоль Вильмса, аниридия, пороки 11 р13 мочеполовой системы, задержка роста и развития) Ангельмана и Прадера-Вилли 15 q 11 -q 12 Рубинштейна-Тейби 16 p 13. 3 Миллера-Дикера 17 p 13. 3 Смита-Мажениса 17 p 11. 2 Ди. Джорджи 22 q 11

Ребенок с синдромом делеции 1 p 36: Микроцефалия, задержка роста, умственная отсталость, эпилепсия, гипотония, прямые брови с глубоко посаженными глазами и гипоплазией средней части лица.
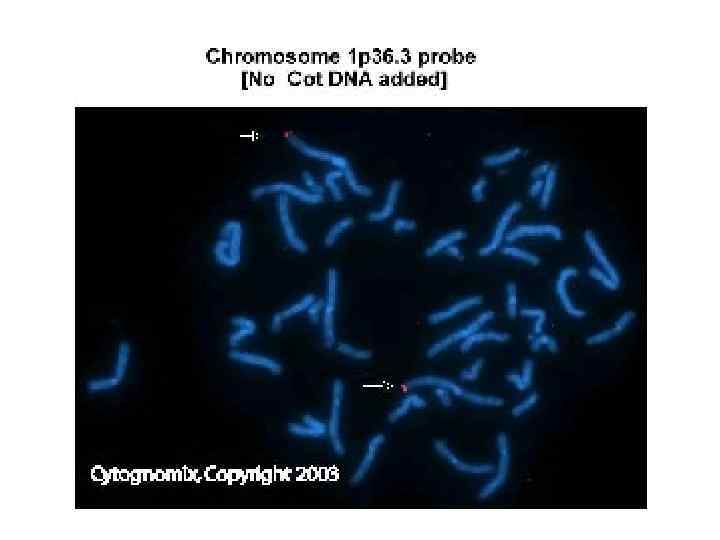
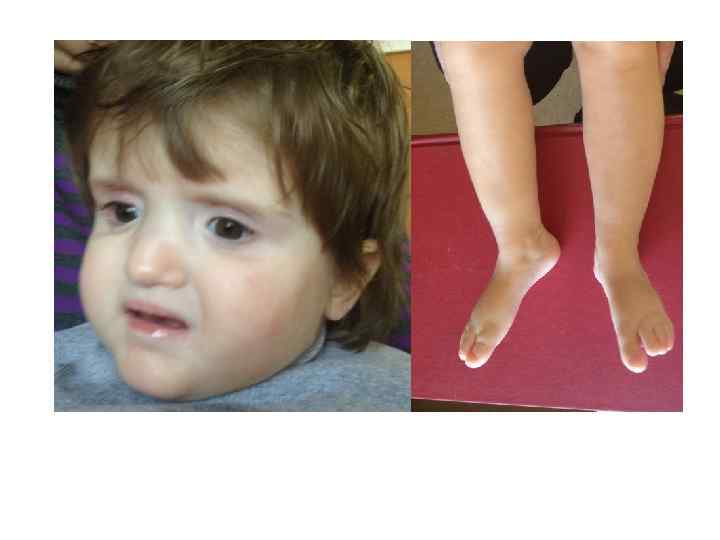
 Синдромы, возникающие вследствие микроделеций Синдромы, возникающие вследствие микродупликаций")
Синдромы реципрокных микроделеций/микродупликаций Участок генома (размер) Синдромы, возникающие вследствие микроделеций Синдромы, возникающие вследствие микродупликаций 7 q 11. 23 (1, 5 – 1, 8 Mb) Синдром Вильямса (OMIM 194050) Синдром микродупликации 7 q 11. 23 (OMIM 609757) 17 р11. 2 (~3, 7 Mb) Синдром Смита-Мажениса (OMIM 182290) Синдром Потоки-Лупски (OMIM 610883) 15 q 11 -q 13 (~4 Mb) Синдром Прадера-Вилли/ Ангельмана (OMIM 17627/ 105830) Синдром микродупликации 15 q 11 -q 13 (OMIM 608636) 17 p 13. 3 (1, 8 -4 Mb) Синдром Миллера-Дикера (OMIM 247200) Синдром микродупликации 17 p 13. 3 (OMIM 613215) 22 q 11. 2 (1, 5 -3 Mb) Синдром Ди Джорджи (OMIM 188400) Синдром микродупликации 22 q 11. 2 (OMIM 608363) Неаллельная гомологичная рекомбинация (NAHR) – механизм рекуррентных микроделеционных/микродупликационных синдромов
 и синдром микродупликации 7 q 11. 23 (OMIM 609757) Результат")
Синдром Вильямса (OMIM 194050) и синдром микродупликации 7 q 11. 23 (OMIM 609757) Результат исследования молекулярного кариотипа: 7 q 11. 23(63, 742, 126 -66, 924, 597)x 1 Результат исследования молекулярного кариотипа: 7 q 11. 23(63, 742, 12666, 924, 597)x 3

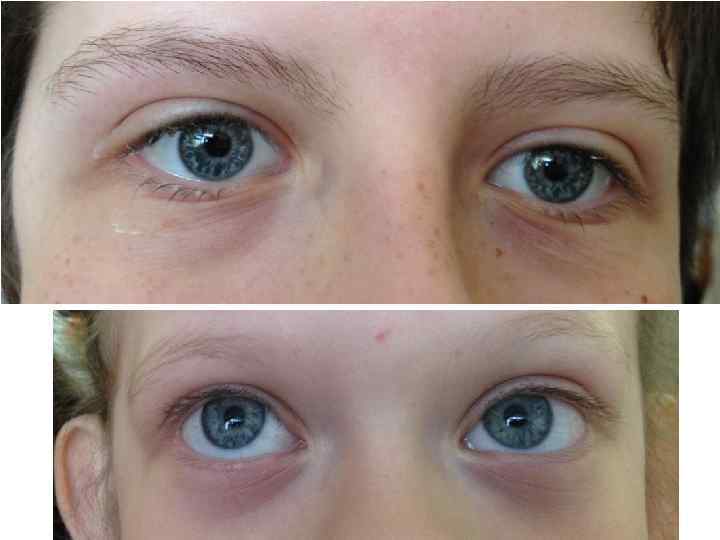
Синдром Вильямса у одного и того же больного в возрастах 7 и 45 лет
 Небольшая задержка роста Умственная отсталость Гиперкальциемия в раннем")
Синдром Вильямса (микроделеция 7 q 11) Небольшая задержка роста Умственная отсталость Гиперкальциемия в раннем детстве Надклапанный стеноз аорты, стеноз легочной артерии Полные щеки, полная нижняя губа ( «лицо эльфа» ) Покатые плечи «Cocktail – party» манера общения в детстве, замкнутость во взрослом возрасте

микроделеция 7 q 11 Норма Синдром Вильямса
")
Синдром Вильямса (OMIM 194050)


 Специфическое лицо:")
Синдром Лангера-Гидеона (трихоринофалангеальный синдром, микроделеция 8 q 34. 11 -q 34. 13) Специфическое лицо: Грушевидный нос Длинный фильтр Гиперплазия нижней челюсти Тонкая верхняя губа Большие оттопыренные ушные раковины Тонкие волосы

Синдром Лангера-Гидеона Конической формы короткие пальцы Множественные хрящевые экзостозы
 опухоль Вильмса, аниридия, пороки мочеполовой системы, задержка роста и")
WAGR синдром (микроделеция 11 р13) опухоль Вильмса, аниридия, пороки мочеполовой системы, задержка роста и развития
")
Интерстициальная делеция короткого плеча хромосомы 11 (отмечена одной стрелкой)
 Умственная отсталость Аутизм Судороги Насильственный смех")
Синдром Ангельмана (микроделеция 15 q 11 -q 12) Умственная отсталость Аутизм Судороги Насильственный смех Атаксия (синдром «счастливой куклы» ) Брахицефалия Макростомия Увеличение нижней челюсти
 и мозаицизма по микроделеции 15 q")
Сочетание делеции гена CDKL 5 (неонатальная эпилептическая энцефалопатия) и мозаицизма по микроделеции 15 q 11. 2 (синдром Ангельмана) Фенотип: сочетание умственной отсталости, аутизма, микробрахицефалии, приступов смеха, «движений механической куклы» , т. е. клинических признаков эпилептической энцефалопатии, связанной с мутациями гена CDKL 5 и синдрома Ангельмана. Результаты исследования молекулярного кариотипа: arr Хр22. 13(18, 519, 70318, 538, 165)× 1, 11 p 13(35, 140, 75535, 377, 738)× 1, 15 q 11. 2(18, 422, 77021, 062, 213)× 1~2
 Умственная отсталость Выраженная гипотония при рождении")
Синдром Прадера-Вилли (микроделеция 15 q 11 -q 12) Умственная отсталость Выраженная гипотония при рождении и впоследствии Ожирение Гипогонадизм Маленькие кисти и стопы

Синдром Прадера-Вилли Особенности фенотипа: У больных с синдромом Прадера-Вилли вследствие унипарентальной дисомии часто встречаются расстройства аутистического спектра и психозы. Генотип: Сегментная потеря гетерозиготности или унипарентальная дисомия длинного плеча хромосомы 15, локализованная в критическом участке синдромов Ангельмана и Прадера. Вилли. Результаты исследования молекулярного кариотипа: arr 15 q 11. 2(24, 251, 567 – 25, 253, 314)x 2 hmz

Синдром Прадера-Вилли Делеции Унипарентальная изодисомия Синдром Ангельмана

Синдром Рубинштейна-Тейби в 13% случаев микроделеции 16 р13. 3 Специфическое лицо: выступающий лоб, дугообразные брови, антимонголоидный разрез глаз, широкая переносица, эпикант, загнутый вниз кончик носа, напоминающая улыбку гримаса, аномалии роста зубов.

Синдром Рубинштейна-Тейби

Синдром Рубинштейна-Тейби Широкие I пальцы кистей и стоп Умственная отсталость глубокая Отставание в росте (у взрослых рост менее 145 -150 см Микроцефалия Пороки сердца: открытый артериальный проток и дефекты перегородок сердца Пороки почек: односторонняя аплазия, удвоение почек, гидронефроз Пороки мозга: агенезия мозолистого тела Крипторхизм
 Выраженая умственная отсталость, судороги, микроцефалия, высокий")
Синдром Миллера-Дикера (синдром лиссенцефалии, микроделеция 17 р13. 3) Выраженая умственная отсталость, судороги, микроцефалия, высокий лоб, суженный в области висков, выступающий затылок, «карпий» рот, маленькая нижняя челюсть, ротированные назад ушные раковины со сглаженным рисунком. На МРТ отсутствие борозд и извилин в больших полушариях головного мозга (лиссенцефалия – гладкий мозг), недоразвитие серого вещества.
 Особенности лица: Брахицефалия Гипоплазия средней части лица")
Синдром Смита-Мажениса (микроделеция 17 p 11. 2) Особенности лица: Брахицефалия Гипоплазия средней части лица Выступающая нижняя челюсть Широкое плоское лицо Вывернутая верхняя губа Близко посаженные глаза Аномалии зубов

Другие признаки синдрома Смита-Мажениса: Когнитивные нарушения Особенности поведения: склонность к аутоагрессии, нарушения сна Короткие широкие кисти Хриплый низкий голос Непостоянные признаки: Задержка роста Сколиоз Аномалии глаз (микрокорнеа – уменьшение размера роговицы, аномалии радужной оболочки и др. ) Снижение слуха Пороки сердца

Микроделеция 17 p 11. 2 при синдроме Смита -Мажениса
 Фенотип: низкий рост, ЗПРР, нарушения сна (частые пробуждения ночью, раннее")
Синдром Смита-Мажениса (OMIM 182290) Фенотип: низкий рост, ЗПРР, нарушения сна (частые пробуждения ночью, раннее просыпание, дневная сонливость), импульсивность, агрессия и аутоагрессия, гипотония мышц, походка на широкой основе, нарушения вскармливания, МАР (выпуклый лоб, монголоидный разрез глаз, гипоплазия средней трети лица, короткий нос с открытыми вперед ноздрями, верхняя губа в форме «шатра» , маленькие кисти и стопы, брахидактилия, плоскостопие) Запись результатов исследования молекулярного кариотипа: arr 17 p 11. 2(16, 758, 204 -20, 393, 335)x 1 Более 90% случаев –интерстициальная делеция 17 p 11. 2 размером 3. 7 -Mb, менее 10% – точковые мутации гена RAI 1 внутри данного региона.
 Синдром Потоки-Лупски (OMIM 610883) Фенотип: низкий рост, ЗПРР, расстройство аутистического")
Синдром Смита-Мажениса (OMIM 182290) Синдром Потоки-Лупски (OMIM 610883) Фенотип: низкий рост, ЗПРР, расстройство аутистического спектра, гипотония мышц, открытое овальное окно, гиподонтия, выступающие лобные бугры, гипоплазия нижней челюсти Результат исследования молекулярного кариотипа: 17 p 11. 2(16, 458, 870 -16, 350)x 3
")
Синдром Ди. Джорджи (делеция 22 q 11)

Синдром Ди-Джорджи • Гипо- или аплазия тимуса, ведущая к нарушениям иммунитета и генералированным инфекциям • Гипоплазия паращитовидных желез, ведущая к гипокальциемическим судорогам у новорожденных • Пороки сердца (тетрада Фалло). • Особенности лица: гипертелоризм или телекант, антимонголоидный разрез глаз, укороченный фильтр, маленькая нижняя челюсть, низко расположенные ушные раковины

Метод молекулярного кариотипирования – прорыв в исследовании микроделеционных синдромов Лаборатория молекулярной цитогенетики НИИ педиатрии и детской хирургии нервно -психических заболеваний Лаборатория цитогенетики и и геномики психических заболеваний Научного центра психического здоровья
 Фенотип: Задержка психомоторного и речевого развития, частые")
Синдром микродупликации гена MECP 2 (OMIM 300260) Фенотип: Задержка психомоторного и речевого развития, частые респираторные инфекции, крипторхизм, микробрахицефалия, широкое лицо, эпикант, гипоплазия средней части лица, заостренный нос, маленький полуоткрытый рот, крупные низко расположенные ушные раковины Генотип пробанда: arr Xq 28(153, 130, 00