
Миопатия эрба код мкб

Связанные заболевания и их лечение
Описания заболеваний
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
Названия
Название: Прогрессирующая мышечная дистрофия Эрба-Рота.
Прогрессирующая мышечная дистрофия Эрба-Рота
Описание
Прогрессирующая мышечная дистрофия Эрба. Рота – аутосомно – рецессивная наследственная миодистрофия, отличающаяся полиморфизмом клинических проявлений и вариативностью скорости прогрессирования симптомов. Может носить нисходящий характер, т. Е. Начинаться со слабости в проксимальных отделах рук, но чаще имеет стандартный восходящий тип распространения мышечных изменений. Диагностика осуществляется методами неврологического осмотра, электрофизиологического тестирования, генеалогического исследования и генетического анализа, по показаниям проводится гистологический анализ биоптата мышечной ткани. Лечение только симптоматическое, позволяющее лишь продлить двигательную активность пациентов. Итог заболевания — полная обездвиженность.
Дополнительные факты
Прогрессирующая мышечная дистрофия Эрба-Рота – самый полиморфный вариант наследственной миодистрофии. От других видов прогрессирующей мышечной дистрофии вариант Эрба-Рота отличается вариабельностью как времени дебюта заболевания, так его клинической картины и течения. Заболевание описано в 1882 г. Немецким неврологом Вильгельмом Эрбом. Одновременно в России изучением этой патологии занимался В. К. Рот, для обозначения миодистрофии он ввел термин «мышечная сухотка». В современной мировой и отечественной неврологии в честь этих исследователей употребляется название «прогрессирующая мышечная дистрофия Эрба-Рота». Частота встречаемости заболевания находится в пределах от 1,5 до 2,5 случаев на 100 тыс. Населения.
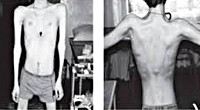
Прогрессирующая мышечная дистрофия Эрба-Рота
Причины
Субстратом дистрофии Эрба-Рота являются патологические метаболические и структурные изменения в мышечной ткани (миопатия). Они возникают в результате генетических мутаций, приводящих к недостатку или полному прекращению синтеза белков, являющихся необходимыми структурными компонентами миоцитов. На сегодняшний день генетике известно не менее 9 хромосомных локусов, аберрации в которых приводят к развитию миодистрофии Эрба-Рота. Чаще всего наблюдаются мутации в локусах 15q15-q21. 1, 13q.
Около 30% генных аномалий возникают de novo, остальные носят семейный характер. Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно. Болеют как мальчики, так и девочки. Патология проявляется, если ребенок получает аномальный ген от каждого из родителей. В случае, когда оба родителя выступают носителями аберрантного гена, но сами не болеют, вероятность развития миодистрофии у ребенка составляет 25%.
Симптомы
Прогрессирующая мышечная дистрофия Эрба-Рота манифестирует в среднем в возрасте 13-16 лет. Однако известны отдельные случаи дебюта болезни в раннем детском возрасте и в возрасте после 20 лет. Мышечная слабость и атрофии возникают в первую очередь в мускулатуре тазового пояса и проксимальных отделов ног. Отмечаются затруднения ходьбы по лестнице, подъема из положения присев на корточки. Типичен симптом Говерса — если пациент сидел на полу, то для того, чтобы подняться, он использует собственное тело как опору.
Со временем дистрофические изменения распространяются на мышечные группы туловища и рук. Такой тип распространения миодистрофии носит название восходящий. Он наиболее типичен для большинства наследственных мышечных дистрофий. Однако в некоторых случаях дистрофии Эрба-Рота наблюдается нисходящий тип распространения патологического процесса, когда мышечная слабость возникает вначале в руках, затем в тазовом поясе, а через несколько лет в мышцах ног.
Тотальная гипотрофия мышц туловища приводит к тому, что у пациентов начинают выступать лопатки (т. Н. «крыловидные лопатки»), талия становиться очень тонкой (т. Н. «осиная талия»), усиливается поясничный лордоз, живот выпячивается вперед. Характерен симптом свободных надплечий — при попытке приподнять больного вверх, удерживая его за подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними. Поражение лицевых мышц влечет за собой гипомимию (т. Н. «лицо сфинкса»), неполное закрытие век, выворачивание и утолщение губ.
Ассоциированные симптомы: Запор. Слабость в ногах. Слабость в руках. Слабость мышц (парез).
Диагностика
Миодистрофия Эрба-Рота диагностируется неврологом и генетиком при сопоставлении данных анамнеза (возраст дебюта заболевания, последовательность развития симптомов), неврологического статуса пациента, ЭФИ нервно-мышечной системы, генеалогических данных, результатов анализа ДНК и микроскопического исследования мышечной ткани.
Дифференциальная диагностика
Дифференцировать миодистрофию Эрба-Рота приходится от других форм этого заболевания (прогрессирующей дистрофии Дюшенна, миодистрофии Дрейфуса и Беккера), дерматомиозита, полимиозита, бокового амиотрофического склероза, токсической миопатии и тд.
При неврологическом осмотре обращает на себя внимание снижение мышечной силы в мускулатуре проксимальных отделов ног и рук, гипотония и гипотрофия указанных мышц, гипорефлексия или полное выпадение локтевых и коленных рефлексов, сохранность всех видов чувствительности. ЭМГ и ЭНГ свидетельствуют о первичном поражении мышечной ткани при сохранности проведения импульсов по нервным стволам.
Генеалогическое исследование подтверждает аутосомно-рецессивный характер наследования. Исследование ДНК может выявить наличие генных мутаций. Однако отрицательный результат исследования не опровергает диагноз, поскольку не всякая мутация может быть обнаружена. Отрицательный анализ ДНК является показанием к биопсии мышц. В биоптате обнаруживаются различные по толщине мышечные волокна, уменьшенное число мышечных ядер, некротические и склеротические изменения.
Резкое повышение уровня креатинфосфокиназы характерно для начального периода дистрофии Эрба-Рота, затем происходит постепенное понижение этого показателя вплоть до нормальных цифр. Обзорная рентгенография грудной клетки позволяет выявить расширение границ сердца, наличие воспалительных изменений легочной ткани. ЭКГ зачастую определяет аритмию и нарушения проводимости. При помощи УЗИ сердца можно диагностировать кардиомиопатию. Для оценки степени сердечных нарушений требуется консультация кардиолога, при подозрении на пневмонию — консультация пульмонолога.
Лечение
Этиопатогенетическая терапия пока не разработана. Симптоматическое лечение направлено на как можно более длительное сохранение двигательной способности пациента. С этой целью применяют медикаментозные курсы, включающие АТФ, витамины Е и группы В, тиоктовую кислоту и тд Занятия лечебной физкультурой должны проводиться ежедневно и включать упражнения на все группы мышц. Регулярно назначаются курсы массажа и физиопроцедуры.
При поражении сердечной мышцы рекомендован инозин, сердечные гликозиды, антиаритмики. При развитии контрактур может потребоваться ортопедическое лечение. Выраженное снижение жизненной емкости легких из-за атрофии дыхательных мышц служит показанием к ИВЛ.
Источник
Прогрессирующая мышечная дистрофия Эрба-Рота является одной из наследственных форм поражения мышечной ткани.
Начало заболевания обычно в возрасте 10-20 лет (возможен дебют до 40 лет); проходит около 10-15 лет до полного обездвиживания. Передача признака – по рецессивному типу, сцепленному с полом.
Информация для врачей. Мышечная дистрофия Эрба-Рота шифруется по МКБ 10 под кодом G71.0. При этом в диагнозе обязательно указывается стадия заболевания (1 – умеренные нарушения движений, 2 – затруднения возникают при ходьбе, выполнении легкой физической работе, 3 – параличи, контрактуры и т.д.). После этого указывается выраженность сопутствующих проявлений (снижение интеллекта, осложнений со стороны сердца), темп прогрессирования (быстрое, среднее или медленное). При утрате способности к передвижению обязательно указывается этот факт.
Причины
Единой точки рения на возникновение мышечных атрофий не существует. Доминирует наследственная теория. Точные биохимические механизмы патогенеза до конца не раскрыты.
Симптомы
Симптомы заболевания вначале не специфичны и включают в себя общую слабость, слабость мышц спины. Постепенно заболевание прогрессирует. Пациент перестает удерживать спину в нормальном положении, развивается гиперлордоз – переразгибание поясницы кзади.
Достаточно рано изменяется походка. Она становится похожей на «утиную» – переваливание ног из-за слабости мышц бедра и тазового пояса. Быстро развивается гипотрофия мышц верхнего плечевого пояса. Также постепенно развивается общая гипотрофия, а затем и атрофия мышц. Иногда имеет псевдогипертрофия голеней – замена мышечной массы жировой и соединительной тканью.
Со временем пациент перестает выполнять многие активные движения. Значимо затрудняется вставание, больным приходится вставать на четвереньки, помогать руками при вставании. Лицо пациента становится амимично, неполностью смыкаются веки, губы же, напротив, выворачиваются кпереди и нередко утолщаются (губы тапира). Мимику такого пациента ещё иногда называют лицом миопата.
Диагностика
Диагноз заболевания обычно выставляется достаточно точно. При постановке диагноза дистрофия Эрба-Рота обращают внимание на возраст в дебюте заболевания, наследственность, скорость прогрессии процесса. В неврологическом осмотре выявляется снижение рефлексов вплоть до выпадения, снижение тонуса мышц, наличие контрактур суставов (из-за неравномерного процесса атрофии мышц).
Вопреки заблуждениям, фасцикулярных подергиваний мышц не бывает. При регистрации биотоков мышц снижается амплитуда, но не частота разрядов. По ЭНМГ определяется укорочение длительности потенциалов действия, полифазность записи.
Биохимически нередко обнаруживается изменение активности креатиникиназы, АСТ и других ферментов. Иногда обнаруживается изменение состава электролитов крови.
Достоверным считают диагноз при проведении гистологического исследования мышц. Меняются форма и размеры мышечных волокон, изменяется восприятие их окраски гистологическими красителями, мышцы перерождаются, увеличивается объем мышечных ядер. Между мышечными волокнами определяется жир, соединительная ткань. При этом отсутствует пучковость распределение волокон, характерная для нейрогенных миопатий.
Лечение
Патогенетической терапии не разработана. Лечение симптоматическое и направлено на уменьшение скорости прогрессирования. Активно используют витамины группы В, витамин Е, АТФ, экстракт алоэ внутримышечно, АТФ. Некоторое время назад использовался анаболический гормон ретаболил, однако часто отмечалось усиление распада мышечной ткани. Также применяют такие препараты, как тиоктовая кислота, рибоксин, актовегин.
Важная роль отводится немедикаментозным методикам воздействия. Массаж пациентам с дистрофией Эрба-Рота должен проводиться в легком темпе, направлен на борьбу с мышечным спазмом, укрепление мышц. Также важная роль принадлежит ЛФК. ЛФК при заболевании должна быть умеренной, но регулярной, в идеале ежедневной. Тренируются все группы мышц.
Постоянство проведение профилактических мероприятий позволяет длительно сохранять возможность больных к самообслуживанию. В моей практике вспоминается пациентка, которая при дебюте заболевания в 25 лет до 60 сохранила способность к самостоятельному передвижению, самообслуживанию, пускай с ограничением.
Прогноз
Прогноз при всех мышечных дистрофиях, как правило, неблагоприятный. Заболевание постепенно прогрессирует, охватывая все группы мышц. Рано или поздно наступает обездвиживание пациента. Хотя при этом само заболевание практически не приводит к смерти пациента. Смерть обусловлена пролежнями, инфекциями легких, мочевыводящих путей и т.п.
Источник
Рубрика МКБ-10: G72.9
МКБ-10 / G00-G99 КЛАСС VI Болезни нервной системы / G70-G73 Болезни нервно-мышечного синапса и мышц / G72 Другие миопатии
Определение и общие сведения[править]
Миопатии — группа наследственных заболеваний, проявляющихся мышечной слабостью и атрофией мышц. Прогрессирующие миопатии называют также миодистрофиями. Гистологически выявляются снижение числа мышечных волокон и вариабельность размеров оставшихся. При амиотрофиях (атрофиях мышц при поражениях двигательных нервов или мотонейронов) патологоанатомическая картина иная. Существует несколько типов миопатий (см. табл. 16.2).
Тяжесть и скорость прогрессирования заболевания зависит от типа миопатии и индивидуальных особенностей. Наиболее тяжело протекает миопатия Дюшенна, при которой большинство больных не доживают до 20 лет. При других миопатиях больные достигают зрелого возраста. Существуют легкие формы миопатий, поражающие лишь определенные группы мышц и практически не ограничивающие жизнедеятельность.
Этиология и патогенез[править]
Патогенез большинства миопатий остается неизвестным. Недавно показано, что в основе этих заболеваний может лежать дефект мембраны мышечных клеток. Миопатии Дюшенна и Беккера связаны с делецией в локусе Xp21 (на коротком плече X-хромосомы). При миопатии Дюшенна в мембране мышечных клеток отсутствует продукт данного гена — дистрофин, а при более доброкачественной миопатии Беккера наблюдается снижение содержания дистрофина либо выявляется дистрофин с аномальным молекулярным весом. Патогенез других миопатий менее изучен.
Клинические проявления[править]
Миопатия неуточненная: Диагностика[править]
1. Выявление в анамнезе и при осмотре у детей и подростков прогрессирующей слабости и похудания мышц позволяет заподозрить миопатию. Однако по клиническим данным невозможно полностью отличить миопатию от амиотрофии. Различные типы миопатий отличаются друг от друга преимущественным поражением тех или иных мышечных групп, скоростью прогрессирования и другими клиническими проявлениями (см. табл. 16.2). В табл. 16.3 приведены признаки, позволяющие отличить амиотрофии от миопатий.
2. Биопсия мышц подтверждает диагноз миопатии и позволяет отличить тяжелые прогрессирующие миопатии от доброкачественных непрогрессирующих (например, от болезни центрального стержня или немалиновой миопатии). Биоптат берут из пораженной, но не наиболее ослабленной мышцы. Чаще всего при миопатиях исследуют дельтовидную и икроножную мышцы, хотя целесообразней проводить биопсию прямой мышцы живота — это не нарушает двигательную активность больного и не приводит к дальнейшему нарастанию слабости вследствие бездействия.
3. Исследование ДНК лейкоцитов с помощью ПЦР выявляет дефект у 70% больных с миопатией Дюшенна и Беккера. Данным методом можно также исследовать культуру ткани плода. Исследование дистрофина в мышечном биоптате позволяет отличить миопатии Дюшенна и Беккера друг от друга и от других миопатий, при которых содержание и структура дистрофина не меняются. Хотя сочетание этих двух методов значительно повышает точность диагноза, в большинстве случаев биопсию не проводят, если при исследовании ДНК лейкоцитов обнаружена характерная делеция.
Дифференциальный диагноз[править]
Миопатия неуточненная: Лечение[править]
1. В легких случаях лечение, особенно на ранней стадии, не требуется; достаточно повторных (каждые 6—12 мес) исследований двигательной функции.
2. В тяжелых случаях, особенно при миопатии Дюшенна, необходимо разъяснить родственникам суть и прогноз заболевания и заручиться их поддержкой.
а. Скрупулезное выполнение назначений врача способно продлить период относительной независимости больного, и семья должна это хорошо понимать. По мере прогрессирования заболевания часто наступает разочарование, однако и в этой ситуации важно поддерживать надежду на стабилизацию процесса и настаивать на продолжении физических упражнений.
б. Поскольку большую часть больных с миопатиями составляют дети, необходимо предусмотреть возможность получения образования и общения со сверстниками. Пока ребенок может без особого труда подниматься по лестнице, он должен посещать обычную школу. В то же время при миопатии Дюшенна интеллект часто снижается (IQ у многих больных составляет менее 90). В таких случаях детей лучше сразу направлять в специальную школу.
в. Цель лечения состоит не только в том, чтобы сохранить способность к самостоятельному передвижению. Болезнь не должна заполнять все существование больного — следует стремиться к тому, чтобы он как можно дольше вел нормальный образ жизни.
3. Преднизон (0,75 мг/кг/сут) способствует увеличению мышечной силы при миопатии Дюшенна. Однако его польза не перевешивает риск побочных эффектов, и поэтому его обычно не назначают. Исключение составляют случаи, когда в результате пневмонии или ателектаза развиваются острые дыхательные расстройства. Чтобы избежать прибавки в весе, во время лечения назначают диету с низким содержанием жиров и соли. При возможном контакте с вирусом varicella-zoster больным, принимающим кортикостероиды, назначают иммуноглобулин против вируса varicella-zoster.
4. ЛФК
а. Тренировки начинают как можно раньше. Больного и его родных следует обучить комплексам упражнений. ЛФК более эффективна, если ее начинают до появления контрактур и деформаций.
б. Цель ЛФК — обеспечить функционирование опорных суставов и предотвратить контрактуры. Так как сгибатели поражаются в несколько меньшей степени, чем разгибатели, контрактуры (в тазобедренных, локтевых и коленных суставах) обычно носят сгибательный характер.
в. Для профилактики контрактур, затрудняющих передвижение и усложняющих уход за больным, необходимы упражнения на объем движений, коррекция положения тела в кровати и кресле, частая смена позы, раннее применение шин.
г. Хотя нет доказательств, что ЛФК замедляет прогрессирование болезни, она тем не менее позволяет отсрочить на несколько лет обездвиженность.
5. Лечение дыхательных расстройств
а. В тяжелых случаях генерализованной миопатии и при поражении мышц гортани и глотки возникают нарушения глотания и дыхания. Дыхательная недостаточность обычно нарастает постепенно и становится явной лишь на поздней стадии.
б. При исследовании функции внешнего дыхания часто выявляются нарушения даже в отсутствие явных дыхательных расстройств. Целесообразно тренировать диафрагмальное дыхание (надувать игрушки или играть на духовых инструментах). Необходимы и специальные дыхательные упражнения под контролем инструктора.
в. На поздней стадии приходится прибегать к ИВЛ и постуральному дренажу.
г. Гиперкапния в отсутствие пневмонии — плохой прогностический признак, поскольку 80% больных с тяжелой миопатией погибает от дыхательной недостаточности. К интубации трахеи при миопатиях прибегают редко.
6. Поддержание подвижности
а. Уменьшение избыточного веса улучшает двигательные возможности и предотвращает гиповентиляцию.
б. Целесообразны ежедневные пешие прогулки продолжительностью не менее 3 ч. Если больной не может ходить, то ему рекомендуют стоять в общей сложности 3 ч в сутки (по 30 мин каждые 3—4 ч).
в. По мере прогрессирования заболевания при ходьбе можно использовать костыли и другие ортопедические приспособления. К передвижению в кресле-каталке переходят как можно позже.
г. При острых сопутствующих заболеваниях слабость может усиливаться, однако постельный режим обычно противопоказан, так как он приводит к еще большему нарастанию мышечной слабости и некоторые больные больше уже никогда не встают.
Профилактика[править]
Профилактика и медико-генетическое консультирование. Если тип наследования заболевания известен, можно предсказать вероятность его развития у ребенка. Больным с миопатиями следует обязательно сообщать степень риска для их будущего потомства.
а. Сцепленные с полом рецессивные заболевания (миопатии Дюшенна и Беккера)
1) Наследование. Вероятность рождения у носителя патологического гена больного сына или дочери-носительницы составляет 50%. Однако примерно у трети больных заболевание в семейном анамнезе отсутствует.
2) Выявление носителей. Примерно у половины носителей в сыворотке увеличена активность КФК. Поскольку активность КФК может колебаться, исследование нужно проводить по меньшей мере 3 раза с интервалом в 10 сут. Накануне взятия крови ограничивают нагрузки, так как они приводят к повышению КФК. Нормальные результаты не исключают возможность носительства. В 60—65% семей с миопатиями Дюшенна или Беккера выявляются характерные мутации в виде интрагенных делеций гена дистрофина. Носительниц в семье больного мальчика можно выявить путем обнаружения делеции в одном из двух аллелей, кодирующих дистрофин. В других случаях используют метод, основанный на анализе полиморфизма длин рестрикционных фрагментов. Точность выявления носительства у девочек достигает 85—90%.
3) Профилактика. Носители могут предотвратить рождение больного ребенка с помощью нескольких способов.
а) Добровольная стерилизация или контрацепция.
б) Пренатальные определение пола и диагностика позволяют произвести аборт по медицинским показаниям.
4) Спонтанная мутация лежит в основе трети случаев миопатии Дюшенна. Чем больше здоровых детей родилось в семье до появления больного ребенка, тем меньше вероятность, что мать является носителем. В этом случае вероятность рождения у нее еще одного больного ребенка невелика.
б. Аутосомно-доминантные заболевания (плече-лопаточно-лицевая миопатия, поздняя дистальная миопатия)
1) Наследование. В типичном случае плече-лопаточно-лицевая дистрофия передается по аутосомно-доминантному типу, однако существуют другие формы с аналогичной клинической картиной, которые наследуются по аутосомно-рецессивному или сцепленному с полом рецессивному механизму. Тяжесть заболевания варьирует. При аутосомно-доминантном типе наследования вероятность передачи заболевания потомству составляет 50%.
2) Носительства не бывает.
3) Профилактика возможна с помощью контрацепции. Если болен мужчина, то для появления здорового потомства можно прибегнуть к искусственному оплодотворению.
в. Аутосомно-рецессивные заболевания (тазо-плечевая миопатия)
1) Наследование. Если оба родителя являются носителями, то один из четырех детей будет болен, два из четырех будут носителями и только один будет полностью здоров.
2) Профилактика: контрацепция и воздержание от близкородственных браков.
Прочее[править]
Источники (ссылки)[править]
Дополнительная литература (рекомендуемая)[править]
1. Bieber, F. R., Hoffman, E. P., and Amos, J. A. Dystrophin analysis in Duchenne muscular dystrophy: Use in fetal diagnosis and in genetic counseling. Am. J. Hum. Genet. 45:362, 1989.
2. Fenichel, G. M., et al. Long term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurology 41:1874, 1991.
3. Hardiman, O., et al. Neuropathic findings in oculopharyngeal muscular dystrophy: A report of seven cases and a review of the literature. Arch. Neurol. 50(5):481, 1993.
4. Harris, S. E., and Cherry, D. B. Childhood progressive muscular dystrophy and the role of physical therapy. Phys. Ther. 54:4, 1974.
5. Hook, R., Anderson, E. F., and Noto, P. Anesthetic management of a parturient with myotonia atrophica. Anesthesiology 43:689, 1975.
6. Iannaccone, S. T. Current status of Duchenne muscular dystrophy. Pediatr. Clin. North Am. 39(4):879, 1992.
7. Love, D. R., et al. Dystrophin and dystrophin-related proteins: A review of protein and RNA studies. Neuromuscul. Disord. 3(1):5, 1993.
8. Miller, G., and Wessel, H. B. Diagnosis of dystrophinopathies: Review for the clinician. Pediatr. Neurol. 9(1):3, 1993.
9. Sarnat, H. B., O’Connor, T., and Byrne, P. A. Clinical effects of myotonic dystrophy on pregnancy and the neonate. Arch. Neurol. 33:459, 1976.
10. Wessel, H. B. Dystrophin: A clinical perspective. Pediatr. Neurol. 6(1):3, 1990.
11. Zellweger, M. D., and Ionasecu, V. Myotonic dystrophy and its differential diagnosis. Acta Neurol. Scand. (Suppl. 55) 49:1, 1973.
Действующие вещества[править]
Источник