
Моносомия по ч хромосоме синдром

Автор venerolog-ginekolog На чтение 5 мин. Опубликовано 18.03.2018
 Хромосомы, отвечающие за пол, обладают особенным свойством. Если нарушения в прочих частях ядра чреваты внутриутробной гибелью плода или тяжелыми патологиями, то при изменении количества половых хромосом возможно рождение вполне жизнеспособного ребенка. В зависимости от вариации набора различается и дальнейшее развитие. Например, многие обладательницы вида моносомии – трисомии по Х-хромосоме или носители кариотипа XYY даже не догадываются о своей необычности. В других случаях улучшить качество жизни помогает современная терапия.
Хромосомы, отвечающие за пол, обладают особенным свойством. Если нарушения в прочих частях ядра чреваты внутриутробной гибелью плода или тяжелыми патологиями, то при изменении количества половых хромосом возможно рождение вполне жизнеспособного ребенка. В зависимости от вариации набора различается и дальнейшее развитие. Например, многие обладательницы вида моносомии – трисомии по Х-хромосоме или носители кариотипа XYY даже не догадываются о своей необычности. В других случаях улучшить качество жизни помогает современная терапия.
Девочкам с единственной Х-хромосомой, несмотря на их особенность, медицина тоже дает шанс преодолеть все трудности, связанные с синдромом моносомии. Но некоторые ограничения в настоящий момент по-прежнему непреодолимы.
Моносомия хромосомы Х: понятие
Моносомия X-хромосомы – генетическое нарушение, также известное как синдром Шерешевского-Тернера. Такое название оно получило по фамилиям исследователей, впервые давших точное описание этой патологии в начале ХХ века. Советский доктор наук Н.А. Шерешевский занимался эндокринологией и считал это состояние болезнью, вызываемой недостаточным развитием половых желез и гипофиза. Но в 1959 году британский генетик Чарльз Форд выявил хромосомную природу происхождения.
В зарубежной литературе иногда используют термин “синдром Ульриха-Тернера” в честь еще одного ученого, занимавшегося исследованием этой проблемы.
Люди с синдромом Шерешевского-Тернера (СШТ) обладают набором из 45 хромосом вместо обычных 46. Их кариотип выглядит как 45 X0 и носит в какой-то степени уникальный характер. Никакая другая моносомия не дает жизнеспособный человеческий организм, способный к относительно стабильному развитию. Все эмбрионы с нехваткой аутосомных хромосом или кариотипом Y0 гибнут в первые месяцы внутриутробного периода.
Разновидность моносомиии
Как и при других генетических патологиях, эта мутация может иметь мозаичную форму. Более того, ее уровень является одним из самых высоких среди других генетических нарушений – около 40%. В этом случае внешние проявления могут быть значительно сглажены или практически отсутствовать, в зависимости от того, какой объем клеток в организме имеет аномальный хромосомный набор. Некоторые из носителей узнают о своей особенности только после кариотипирования при проблемах с зачатием.

Существуют следующие варианты мозаицизма:
- кариотип 45X/46XX – в организме присутствуют клетки сразу с нормальным и аномальным женским набором;
- кариотип 46 Х Хр- и 46 X Xq– – отсутствие части хромосомы или так называемая делеция плеч;
- кариотип 46 Xi(Xq) или 46,X,i(Xp) – наличие изохромосомы с идентичными плечами;
- кариотип 45 Х/46 XY – в организме присутствует мужская половая хромосома, способная дать частичное развитие соответствующего типа, но в большинстве случаев гениталии амбисексуальны – в брюшной полости одновременно находится неопустившееся яичко и зачаточная матка.
По каким причинам происходит патология
Причина моносомии кроется в мутации, которая возникает спонтанно при образовании плодного яйца в первые дни беременности. Одна из родительских Х-хромосом (в большинстве случаев отцовская) частично повреждается или вовсе выпадает из процесса формирования кариотипа.
В отличие от многих других генетических аномалий, данное нарушение не зависит от возраста родителей.
Протекание беременности при СШТ у плода почти всегда сопровождается угрозой прерывания. Возможны преждевременные роды.
Симптомы
 Визуальные признаки нарушения (специалисты называют их стигмами) часто заметны уже в первые дни жизни ребенка. Девочки с СШТ с самого рождения обладают маленькими размерами даже при доношенной беременности. Рост новорожденных не превышает 48 см, а вес колеблется в пределах 2800 г, у них наблюдается лимфостаз, выражающийся в отеках конечностей, и специфичное строение шеи. Более глубокое обследование выявляет пороки развития внутренних органов, главным образом сердечно-сосудистой и мочеполовой систем.
Визуальные признаки нарушения (специалисты называют их стигмами) часто заметны уже в первые дни жизни ребенка. Девочки с СШТ с самого рождения обладают маленькими размерами даже при доношенной беременности. Рост новорожденных не превышает 48 см, а вес колеблется в пределах 2800 г, у них наблюдается лимфостаз, выражающийся в отеках конечностей, и специфичное строение шеи. Более глубокое обследование выявляет пороки развития внутренних органов, главным образом сердечно-сосудистой и мочеполовой систем.
По мере взросления организма формируются более явные признаки синдрома:
- малый рост, редко превышающий 145 см;
- непропорциональное телосложение: короткая шея, иногда имеющая клиновидные складки, бочкообразная грудная клетка, деформация локтевых суставов;
- низкая плотность костной ткани и угроза развития остеопороза;
- недоразвитие внешних половых органов и молочных желез;
- многочисленные родинки и другие кожные образования;
- в психологическом состоянии наблюдается инфантилизм при успешной социальной адаптации.
Методы диагностики
СШТ можно выявить пренатально, но в большинстве случаев нарушение диагностируется уже после рождения ребенка. Во время беременности материалом для исследования служат ворсины хориона, околоплодная жидкость или пуповинная кровь. У появившихся на свет девочек для генетического анализа берут кровь из вены.
Важно знать всем женщинам: что такое аденоз молочных желез, симптомы и методы лечения заболевания.
Из-за чего возникает гематометра, и каких осложнений опасаться читайте тут.
О прыщах перед месячными: https://venerolog-ginekolog.ru/gynecology/diseases/pryishhi-pered-mesyachnyimi.html.
Лечение и наблюдение пациентов
Полностью ликвидировать проявления синдрома нельзя, но качество жизни улучшает гормональная терапия. Начиная с младшего школьного возраста девочкам показан прием соматотропина для приближения конечного роста к норме. В подростковом возрасте применение гормональных препаратов восполняет недостаток собственного эстрогена, что помогает организму сформироваться по женскому типу – как внешне, так и внутренне.
Вероятность здоровой беременности
Несмотря на то, что заместительная гормональная терапия позволяет вырастить репродуктивную систему до взрослого состояния, истинный СШТ, как правило, сопровождается полным отсутствием собственных яйцеклеток. Однако при мозаичной разновидности патологии есть вероятность их созревания, нарушения гормонального фона проявляются в нестабильном менструальном цикле. Естественная беременность и успешные роды наступают в единичных случаях, чаще всего в этот период требуется гормональная поддержка для предотвращения выкидышей. В большинстве же случаев необходимо использование процедуры экстракорпорального оплодотворения, иногда – с донорской яйцеклеткой.
Если обладательнице мозаичного СШТ удалось забеременеть, вероятность родить здорового ребенка у нее не намного ниже, чем у обычных женщин.
Интересное видео о X-хромосоме:
https://youtu.be/t_GAezyiStY
Источник
У каждого здорового человека имеется 23 пары хромосом с генетическим материалом. Моносомией называют состояние, при котором отсутствует одна хромосома в какой-то паре. Моносомия Х не провоцирует гибель плода при отсутствии одной из хромосом. Это исключительно женская патология, когда генотип обозначают 44 Х0, а не стандартными 44 ХХ.

Обычно моносомия отмечается только в соматических клетках, если же патология затрагивает половые хромосомы, ее называют аномалией по Х хромосоме, моносомией Х или синдромом Шерешевского-Тернера. Заболевание встречается у 5 девочек из 10 тысяч новорожденных.
Причины возникновения
Точные причины моносомии Х не определены, но механизмы развития изучены хорошо. В половине случаев синдром связывают с тем, что Х-хромосома не разошлась при образовании половых клеток. При формировании половых клеток набор хромосом их предшественников расходится по разным клеткам. Если этот процесс нарушится, в одну яйцеклетку может попасть обе Х-хромосомы, а другая будет «пустой». Такой клетке дают обозначение 22 00 (вместо 22 ХХ).
При оплодотворении «пустой» яйцеклетки есть два варианта. Либо зигота будет иметь набор 44 Х0, а новорожденная девочка унаследует синдром, либо зигота получит Y-хромосому от отца и будет иметь набор 44 Y0, с которым зародыш развиваться не может, ведь от Х-хромосомы он получает необходимые белки.
Во всех остальных случаях образование половых клеток происходит без проблем, яйцеклетки имеют полный набор 22 Х и после оплодотворения образуют необходимые клетки 44 ХХ и 44 YY. На раннем этапе развития плода возможно неправильное деление клеток и потеря одной половой хромосомы. Это называется феноменом мозаицизма или частичной моносомии: часть клеток будет иметь моносомию Х, а другая делится без потерь.
Результатом патологии становятся нарушения в строении половой системы вплоть до гермафродитизма, хотя возможно и нормальное формирование половых органов по присущему типу. Степень нарушений зависит от того, какая доля клеточного материала утратила генетическую информацию.
Виды моносомии Х
При полной моносомии Х одна из половых хромосом безвозвратно утеряна, чаще всего именно та, которая несет в себе информацию о вторичных половых признаках, половых органах, железах внутренней секреции. У пациентки могут отсутствовать или быть недоразвитыми половые органы, что сказывается на половых гормонах.
При мозаичной моносомии, когда одна половая хромосома нормальная, а вторая «поломана», некоторая информация о развитии передается, но ее слишком мало, чтобы обеспечивать полноценное формирование половой системы.
Риски аномалии
Вероятность рождения ребенка с синдромом Шерешевского-Тернера не связана с возрастом матери. Аномалия в той или иной форме встречается в 1% из всех случаев зачатий, и большинство эмбрионов гибнет до рождения в результате выкидыша или замерзшей беременности. В 10% выкидышей причиной является именно моносомия Х у плода.
Только 1% таких беременностей сохраняется до 7 месяца. Если ребенок рождается жизнеспособным, он может прожить нормальную по длительности жизнь. У большинства таких девочек диагностируют бесплодие, хотя при мозаичной форме вероятность забеременеть выше, чем при истинной.

Диагностика синдрома Шерешевского-Тернера
Распознать симптомы моносомии Х можно во время планового УЗИ плода.
Характерные признаки синдрома Шерешевского-Тернера у плода:
- пороки сердца;
- водянка мозга;
- почечные аномалии;
- кистозная гигрома;
- брахицефалия (короткоголовость);
- признаки задержки внутриутробного развития.
Если имеется один или несколько признаков, необходимо исследовать кариотип, то есть состав и количество хромосом. Это возможно путем амниоцентеза и биопсии хориона. Такие исследования очень информативны и точны, а вероятность ошибки крайне низка. Истинную моносомию Х диагностируют сразу, хотя для верности проводят дифференциальную диагностику с синдромом Нунан.
Вероятность рождения второго ребенка с нарушениями не повышается, но родителям больной девочки рекомендуется все-таки пройти генетическую консультацию. Углубленные генетические исследования позволяют определить изменения в наборе хромосом и вероятность аномалий в развитии.
Симптомы
Человек с аномалией Х-хромосомы имеет аномальное строение тела. Внешние проявления синдрома могут быть очень разнообразными: от слабо выраженных признаков до тяжелых аномалий, которые мешают в жизнедеятельности.
Физиологические признаки:
- широкая, но плоская грудь;
- вертикальные складки кожи на шее;
- смещение челюсти;
- низкое положение ушей;
- опущение границы роста волос на шее;
- плоскостопие;
- деформация конечностей;
- подвижность суставов;
- узкий таз и короткие ноги;
- рост 140-147 см;
- деформация ногтей (короткие и узкие ногтевые пластины, глубокое расположение в ногтевом ложе);
- короткие пальцы;
- искривление локтевых суставов;
- деформации позвоночника (как правило, сколиотические);
- опущение век, косоглазие, рефракционные нарушения.
У людей с моносомией Х часто возникают отеки. Из-за врожденных нарушений страдает мочевыделительная система. Почки могут иметь подковообразную форму и смещаться, удваиваются мочеточники, деформируются почечные вены и артерии. Иногда у девочек диагностируют недоразвитость органов этой системы и различные аномалии строения.
У детей дошкольного возраста периодически возникают отиты и, как следствие, развивается тугоухость. Из-за нарушенного обмена веществ всех видов у женщин с моносомией Х часто выявляют ожирение, облысение или оволосение, гипотиреоз, большое количество родимых пятен, целиакию, сахарный диабет, гипертонию.

Как наблюдают девочек с аномалией Х
Поскольку моносомия Х связана с изменением в генетическом материале, эта болезнь неизлечима. Пациента ставят на учет сначала к педиатру, потом к терапевту, а также к нефрологу, кардиологу, эндокринологу, отоларингологу, стоматологу и другим врачам по показаниям.
В первый год жизни необходимо обследовать зрительную систему. У девочек до 4 лет проверяют состояние тазобедренных суставов, чтобы исключить дисплазию. Нередко требуется коррекция сколиоза, а после 18 лет лечение остеопороза. Оценку функциональности сердца проводят каждые 5 лет (ЭКГ, ЭхоКГ, МРТ). С такой же периодичностью проверяют органы слуха.
До 18 лет необходимо каждый год проверять уровень гормонов щитовидной железы, работу почек и печени. В будущем важно также контролировать уровень холестерина и сахара в крови. Каждые 3 года следует сдавать анализ на антитела к трансглутаминазе, чтобы вовремя диагностировать целиакию.
Влияние на психику и способности человека
При достижении ребенком 4-5 лет обязательно проводят оценку знаний и умений, психического и физического развития. Оценку интеллектуального развития и адаптации в социуме проводят ежегодно.
Интеллектуальные способности у девочек с синдромом Шерешевского-Тернера не снижены, но повышена эмоциональная неустойчивость. Требуется психологическая поддержка, ведь многие страдают из-за низкого роста и половой недоразвитости.
Обучаются такие люди благодаря усидчивости, но приходится бороться с плохой концентрацией внимания и недостатком логического мышления. Для девочек с моносомией Х характера аккуратность, внимание к мелочам в быту, упрямство и приподнятое настроение.
Особенности развития половой системы
Деформация яичников отмечается уже с третьего месяца развития плода, когда соединительная ткань начинает вытеснять зародышевые клетки. При этом у 18% женщин с мозаичной моносомией обнаруживают внешне нормальные органы.
Первичное бесплодие возникает из-за нарушений в репродуктивной системе (недоразвитость яичников, матки и фаллопиевых труб, отсутствие половых желез). В большинстве случаев отмечается задержка полового развития, инфантилизм, аменорея, недостаток эстрогена. Нарушение выработки эстрогенов становится причиной раннего развития остеопороза с частыми переломами.
Вторичные половые признаки появляются у 20% девочек, но только у 5% женщин с моносомией Х проходят менструации. Цикл, как правило, нерегулярный, а менопауза наступает рано.

Можно ли выносить ребенка при моносомии Х
Несмотря на все имеющиеся нарушения, треть женщин с моносомией Х имеют в какой-то степени выраженное половое развитие. Среди них 5% могут выносить и родить ребенка. Нужно учитывать, что женщина с синдромом Шерешевского-Тернера способна передать заболевание своей дочери. У детей таких пациентов повышен риск развития аномалий.
Вопреки тому, что подавляющее большинство считается первично бесплодным, у половины пациенток формируются фолликулы. Это характерно для мозаичной формы моносомии, хотя у 25% женщин даже при истинной (тяжелой) форме синдрома есть фолликулы и яйцеклетки.
Теоретически такие яйцеклетки можно криоконсервировать и использовать в репродуктивной медицине, но при моносомии Х более безопасным считается использование донорской яйцеклетки. Этот способ обеспечивает 30-60% успеха. Важно следить за тем, чтобы был только один зародыш, ведь беременность двойней или тройней при синдроме Шерешевского-Тернера опасна преэклампсией и гестационным диабетом.
Беременность при моносомии Х возможна только при нормальном строении влагалища и матки, поэтому до зачатия нужно пройти обследование и проконсультироваться со специалистами. Тяжелые пороки репродуктивной системы делают беременность невозможной.
Какое лечение необходимо при синдроме Шерешевского-Тернера
Терапия патологии направлена на стимуляцию роста и правильное формирование вторичных половых признаков. Желательно также добиться наступления менструации и становления цикла.
Взрослые и дети с синдромом Шерешевского-Тернера должны придерживаться принципов диетического питания. Из-за низкого роста таким женщинам нужно меньше калорий. Поскольку из-за недостаточности яичников имеется дефицит эстрогена, при моносомии Х нередко развивается остеопороз.

Важно, чтобы девочка получала достаточное количество кальция и витамина D и следила за своим весом. В этом поможет умеренная физическая активность, физкультура и даже некоторые виды спорта.
Отставание в росте отмечается до 3 лет и после 10. Низкорослым девочкам могут назначить инъекции гормона роста и анаболические стероиды. Терапию продолжают до тех пор, пока скорость прибавки длины тела не снизится до 2 см в год. Такое лечение помогает пациенткам вырасти до 150 см, а без гормональной терапии максимум роста при моносомии Х составляет 145 см.
Чтобы имитировать половое развитие, назначают стероидные гормоны. До 12-15 лет только эстрогены, а позже также прогестины. Для удобства пациенток обычно выписывают препараты в форме пластырей. Иногда требуется дополнительный прием мужских половых гормонов.
Патологии сердца требуют приема антибиотиков перед стоматическими и хирургическими процедурами. Так уменьшают вероятность эндокардита. При планировании оперативного вмешательства также нужно учитывать повышенный риск образования келоидных рубцов.
По показаниям могут быть назначены гормоны щитовидной железы, препараты для снижения артериального давления, витаминные добавки. Самое важное, чтобы терапия проводилась совместно, и каждый врач учитывал назначения другого специалиста.
Какие гормональные препараты вы принимали для стимуляции овуляции?
Источник
Оглавление
- Введение
- Краткое содержание
- Аномалии числа хромосом (анеуплоидии)
- Моносомия по X-хромосоме (45,X, или Синдром Шерешевского-Тёрнера)
- 47,XXY Синдром Клайнфельтера
- 47,XYY
- 47,XXX
- Другие заболевания
- Мозаицизм 45,X/46,XX
- Мозаицизм 45,X/46,XY
- Структурные аномалии хромосом
- Изохромосома Xq
- Делеция Xp22.11
- Делеция Xp22.3
- Делеции Xp22 SHOX
- Делеции Xp11.22
- Дупликации Xp.22.31
- Синдром дупликации ME2CP
Введение
Патологии половых хромосом связаны с нарушением их количества (т. е. анеуплоидии, например, моносомия X-хромосомы) или со структурными дефектами (например, такие геномные перестройки, как синдром дупликации гена MECP2). Частота врожденных хромосомных мутаций составляет как минимум 1:400.
Краткое содержание
Патологии половых хромосом могут быть обусловлены нарушением их количества (анеуплоидиями) или же структурными дефектами.
Наиболее распространенные анеуплоидии половых хромосом: 45,X (Синдром Тёрнера); 47,XXY (Синдром Клайнфельтера); 47,XYY; и 47,XXX. Мозаицизм по половым хромосомам с присутствием в организме клеток с нормальным генотипом нередок. Два наиболее распространенных вида мозаицизма половых хромосом — 45,X/46,XX и 45,X/46,XY. Тяжесть фенотипических проявлений у пациентов с мозаицизмом соответствует доле аномальных клеток.
Структурные патологии X- и Y-хромосом прежде всего включают изохромосомы, делеции, дупликации, кольцевые хромосомы и транслокации.
Одним из примеров геномного расстройства является дупликация гена MECP2 у мужчин, выражающаяся в наличии мышечной гипотонии, тяжелой умственной отсталости, задержки речевого развития, нарушения глотания, частых респираторных инфекций, а также судорожных приступов (тонико-клонические судороги, не поддающихся лечению).
Аномалии числа хромосом (анеуплоидии)
Наиболее частыми анеуплоидиями половых хромосом являются 45,X (Синдром Шерешевского-Тернера); 47,XXY (Синдром Клайнфельтера); 47,XYY и 47,XXX с частотой возникновения приблизительно 1/2500, от 1/500 до 1/1000, от 1/900 до 1500 и 1/1000 соответственно. Мозаицизм по половым хромосомам с присутствием в организме клеток с нормальным генотипом нередок. Два наиболее распространенных вида мозаицизма половых хромосом — 45,X/46,XX и 45,X/46,XY. Тяжесть фенотипических проявлений у пациентов с мозаицизмом соответствует проценту аномальных клеток.
Моносомия по X-хромосоме (45,X, или Синдром Шерешевского-Тёрнера)
Большинство пациентов с синдромом Шерешевского-Тёрнера имеют моносомию по Х-хромосоме, кариотип 45,X. Другие формы синдрома включают мозаицизм по хромосоме Х, например, 45,X/46,XX или 45,X/46,XY с частичной делецией Y-хромосомы. У некоторых пациентов имеется структурная аномалия второй X-хромосомы (например, изохромосомия длинного плеча X-хромосомы или делеция короткого плеча). Делеции, включающие в себя дистальную часть короткого плеча Y-хромосомы, также ассоциированы с фенотипом синдрома Тёрнера, поскольку в данном случае у пациентов отсутствуют так называемые анти-тёрнеровские гены (SHOX, RPSY4 и ZFY). Делеции короткого плеча X-хромосомы также связывают с фенотипом синдрома Тёрнера. В большинстве представляют собой единичные случаи.
Синдром Шерешевского-Тёрнера характеризуется низкорослостью и некоторыми из следующих проявлений: дисморфия лица, включающая низко посаженные уши, кожные складки на шее, щитообразная грудная клетка (широкая, с большим расстоянием между сосками), лимфедема, вальгусная деформация локтевого сустава, короткая четвертая пястная кость, гипоплазия ногтевых пластин, пигментные пятна и врожденные пороки сердца. Среди пороков сердца типичными и наиболее часто встречающимся являются дефекты сосудов и коарктация аорты. Вдобавок у пациентов, страдающих синдромом Тёрнера, развиваются полосковидные гонады, наблюдается нарушение овуляции и задержка полового развития. Также встречаются дефекты развития почек (подковообразная почка). Лимфедема нижних отделов конечностей может быть единственным клиническим признаком, наблюдаемым у новорожденных. Лица с синдромом Тёрнера, несущие генетический материал Y-хромосомы, имеют повышенный риск развития гонадобластомы.
47,XXY Синдром Клайнфельтера
Синдром Клайнфельтера является самой распространенной патологией числа половых хромосом, вызывающей первичный гипогонадизм. Кариотип 47,XXY является результатом нерасхождения половых хромосом и может быть как материнским, так и отцовским по происхождению. Большинство случаев болезни обнаруживается постнатально и диагностируется при определении причин бесплодия, выявлении гинекомастии, крипторхизма или же неврологических нарушений.
Рис. Нерасхождение половых хромосом
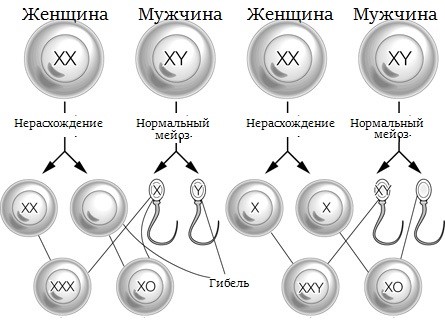
Новорожденные мальчики с кариотипом 47,XXY фенотипически нормальны, с физиологически нормальными мужскими наружными половыми органами и без какой-либо видимой дисморфии. Основные клинические проявления синдрома Клайнфельтера, включающие высокий рост, маленькие яички и бесплодие (азооспермия), становятся выраженными в постпубертатном периоде. У пациентов с синдромом Клайнфельтера повышен риск психических расстройств, расстройств аутистического характера и социальных проблем. У пациентов с диагностированным синдромом Клайнфельтера следует оценивать неврологический статус и направлять к эндокринологу.
47,XYY
Лица с кариотипом 47,XYY имеют высокий рост, у них может отмечаться умеренная задержка в двигательном и речевом развитии. Для многих из них требуется повышенное внимание к обучению, но, как правило, все они учатся в основных общеобразовательных школах. Половое развитие проходит нормально и большинство мальчиков фертильны. Из-за невыраженности фенотипа и отсутствия связанных с этим проблем со здоровьем, многие лица с кариотипом 47,XYY на протяжении всей их жизни остаются недиагностированными.
Ранее сообщалось, что у мужчин с 47,XYY повышена агрессия, что выражается в их агрессивном поведении. Однако последующие крупномасштабные совместные исследования европейских и американских генетиков показали, что статистика повышенной криминальной деятельности мужчин с XYY коррелировала с их низким социально-экономическим статусом по причине низкого значения IQ (около 10 баллов), что приводило к определенным трудностям с законом и, чаще, незначительным правонарушениям. У лиц с 47,XYY отмечаются более высокие показатели синдрома дефицита внимания и гиперактивности, а также расстройств аутистического характера. Таким пациентам рекомендуется оценка их нервно-психического развития, учитывая широкую распространённость трудностей в обучении и поведенческих проблем.
47,XXX
47,XXX (она же трисомия по X-хромосоме) является самой распространенной патологией половых хромосом у женщин. Трисомия по Х-хромосоме диагностируется внутриутробно в ходе генетического скрининга. У женщин с кариотипом 47,XXX нет повышенного риска развития плода с хромосомными аномалиями.
Обследование 155 женщин с кариотипом 47,XXX показало, что 62 процента из них были физически нормальными. Таким образом, для большинства лиц с кариотипом 47,XXX диагноз никогда не устанавливается. У женщин с 47,XXX отмечается высокий рост; (средняя длина окружности головы варьирует в пределах 25 – 35 процентиль, однако к подростковому возрасту для многих может достигать 80 процентиль). Половозрелость и фертильность чаще всего в норме, но может отмечаться преждевременное угасание функции яичников.
В следующем обследовании одиннадцати младенцев с кариотипом 47,XXX было показано, что коэффициент интеллекта девочек с рождения был на 15-20 баллов ниже, чем у их братьев. Поэтому рекомендуется отслеживать задержки в развитии и выявлять наличие психологических проблем в дальнейшем.
Другие заболевания
Сообщалось о более чем ста случаях кариотипа 49,XXXXY, по меньшей мере двадцати случаях 49,XXXXX и нескольких – 49,XYYYY. Прослеживается прямая зависимость между числом дополнительных половых хромосом и тяжестью фенотипических проявлений у пациентов. В исследовании тетра- и пентасомии половых хромосом сделан вывод о том, что полисомия по X-хромосоме связана с более тяжкими последствиями, чем полисомия по Y-хромосоме. Было показано, что уровень интеллекта IQ снижается на 10 пунктов с каждой лишней X-хромосомой от их нормального числа.
49,XXXXY Характерными клиническими чертами кариотипа XXXXY являются запавшая переносица с широким или приподнятым кончиком носа, широко расположенные глаза, веко-носовые складки, скелетные патологии (особенно лучелоктевой синостоз), врожденные сердечные заболевания, эндокринные расстройства и высокая степень гипогонадизма и гипогенитализма. Также обычным являются выраженная умственная отсталость и умеренная низкорослость. Хотя лиц с таким кариотипом часто относят к случаям синдрома Клайнфельтера, все характерные черты XXXXY довольно отчетливо указывают именно на данный фенотип.
49,XXXXX У женщин с кариотипом 49,XXXXX (пентасомия по X-хромосоме) всегда присутствует умственная отсталость. Другие проявления, такие как черпено-лицевые, сердечно-сосудистые и скелетные патологии довольно непостоянны. У пациентов, страдающих пентасомией по X-хромосоме, могут проявляться схожие черты с теми, что наблюдаются при синдроме Дауна. Лучелоктевой синостоз также часто выражен у пациентов с большим числом X-хромосом. Некоторые пациенты имеют мозаицизм 48,XXXX и 49,XXXXX.
Мозаицизм 45,X/46,XX
Это наиболее распространенный мозаицизм половых хромосом, который диагностируется при амниоцентезе и пренатальном кариотипировании. У лиц с данным типом мозаицизма имеются более легкие клинические черты синдрома Тёрнера. Многие женщины прошли половое созревание и смогли воспроизвести потомство.
Из 156 пренатально диагностированных случаев мозаицизма 45,X/46,XX 14% случаев имели ненормальный исход. Было зарегистрировано два мертворождения и 20 случаев ненормального фенотипа (у 12 имелись некоторые черты синдрома Тёрнера, а остальные 8 носили характер аномалий, возможно, не связанных с ним). Более 85 % девочек имели нормальный фенотип при рождении, либо он был установлен по результатам медицинского прерывания беременности. Однако, главные черты синдрома Тёрнера (такие как низкий рост и отсутствие вторичных половых признаков) проявились только в детстве или юности, и не были замечены в младенчестве. У некоторых женщин с нормальным фенотипом, при нарушении функции яичников, выявляется мозаицизм 45,X/46,XX.
Мозаицизм 45,X/46,XY
Мозаицизм с наличием 45,X/46,XY имеет широкий фенотипический спектр. Например, в ретроспективной серии 151 постнатально диагностированных случаев мозаицизма 45,X/46,XY, 42 % пациентов — девочки по фенотипу, с наличием типичного или нетипичного синдрома Тёрнера. Еще у 42 % наблюдались неопределённые наружные половые органы и асимметричные гонады (смешанный гонадный дисгенез), наконец, у 15% был мужской фенотип с неполной маскулинизацией. Таким образом, все случаи, диагностированные постнатально, были фенотипически патологичными. Напротив, среди 80 пренатально диагностированных случаев мозаицизма 45,X/46,XY 74 92,6% были нормальными по фенотипу мальчиками. Это может объяснить тот факт, что дети или взрослые с наличием мозаицизма, но нормальным фенотипом вряд ли стали бы обращаться за медицинской помощью (ошибка обращаемости).
Структурные аномалии хромосом
Структурные патологии включают, прежде всего, изохромосомы, делеции, дупликации, кольцевые хромосомы и транслокации.
Изохромосома Xq
Изохромосома длинного плеча X-хромосомы, isoXq или i(Xq), при наличии которой короткое плечо (p) исключено (отсутствует/редуцировано) и заменено точной копией длинного плеча (q), — является наиболее распространенной аномалией половых хромосом.
Наличие структурной патологии не связывают с повышенным возрастным риском родителей. Изохромосомия 46,X,i(Xq) может быть выражением мозаицизма, когда в организме присутствуют две генетически разные клеточные популяции: нормальная – 46,XX и 45,X.
Изохромосомы Xq и Xy ассоциируют с синдромом Тёрнера, возможно, потому, что главный анти-тёрнеровский ген SHOX располагается на дистальной части коротких плеч X-и Y-хромосом (на псевдоаутосомных областях). Изохромосома Xq также выявляется у пациентов в одной из вариаций синдрома Клайнфельтера, 47,X,i(Xq),Y.
Делеция Xp22.11
Делеция Xp22.11 включает в себя ген PTCHD1. Сообщалось о выявлении в нескольких семьях с расстройствами аутистического характера, а также в трёх семьях с умственной отсталостью. Ген PTCHD1 является геном-кандидатом в отношении Х-сцепленной умственной отсталости, проявляющейся с аутизмом или без аутизма. Функция и роль данного гена неизвестны.
Делеция Xp22.3
Делеция данной области часто ассоциируется с синдромом микрофтальмии и линейных дефектов кожи (MLS) и является Х-сцепленным доминантным нарушением, то есть, летальным для мужчин и поэтому прослеживающимся только у женщин. Ген в данной области кодирует митохондриальную цитохром-c-синтазу (HCCS). Клиническое проявление MLS выражается наличием микрофтальмии и анофтальмии (одно- или двусторонней) и линейными дефектами кожи, в основном лица и шеи, которые со временем проходят. Структурные патологии головного мозга, задержка в развитии и приступы (припадки) тоже входят в состав клинической картины. Нарушения сердечной деятельности (как гипертоническая кардиомиопатия и аритмия), низкий рост, грыжа диафрагмы, ногтевая дистрофия, преаурикулярный свищ, потеря слуха, мочеполовые мальформации (пороки развития, неправильное формирование) также являются частыми клиническими явлениями.
Скрининговая оценка предусматривает офтальмологический и дерматологический осмотр, оценку общего развития, выполнение эхокардиограммы, магнитно-резонансной томографии мозга (МРТ) и электроэнцефалограммы (ЭЭГ).
Делеции Xp22 SHOX
Делеция Xp22 включает в себя ген SHOX, мутация которого является причиной идиопатического низкого роста. Ген SHOX находится в псевдоаутосомном регионе 1 X- и Y-хромосом. Этот ген считается ответственным за низкорослость при синдроме Тёрнера, а гаплонедостаточность данного гена вызывает дисхондростеоз Лери-Вейлля. Дисхондростеоз Лери-Вейлля характеризуется низким ростом, наиболее выражено проявляющимся у женщин, а также хроническим подвывихом кисти (деформацией костей запястья, деформация Маделунга). Гомозиготные делеции гена SHOX вызывают дисплазию Лангера, более тяжелую форму метафизарной дисплазии. Делеции гена SHOX легко обнаруживаются у пациентов с низким ростом, без каких-либо других специфических особенностей в строении их скелета. Более чем 60% перестроек SHOX – это делеции гена; при отсутствии делеций сравнительная геномная гибридизация с последующим секвенированием для выявления и установления точечных мутаций, является клиническим обследованием идиопатического низкого роста.
Делеции Xp11.22
Делеции региона Xp11.22 включают ген PHF8 (кодирует пальцевидный белок PHD8), мутации которого связывают с умственной отсталост