
На каком сроке определить синдром арнольда киари

Синдром Арнольда – Киари – это совокупность изменений в ЦНС плода, возникающих в результате порока развития продолговатого мозга и обусловленных асинхронным ростом ствола мозга и спинного мозга. Впервые он был описан Н. Chiari в 1896 г. Это состояние характеризуется каудальным смещением продолговатого мозга, моста и червя мозжечка, когда все эти структуры оказываются в шейной части позвоночника.
Выделяют три типа мальформации Арнольда-Киари в зависимости от степени вклинения структур продолговатого мозга в позвоночник:
I тип характеризуется удлинением ствола мозга и проникновением миндалин мозжечка в шейный отдел позвоночного канала;
II тип – вклинивание дисплазированного мозжечка в большое затылочное отверстие в сочетании с удлинением ствола мозга;
III тип – тотальное смещение структур заднего мозга в расширенное затылочное отверстие, сопровождаемое грыжей в затылочной области.
Истинная частота различных типов синдрома Арнольда – Киари, да и частота этого порока в целом, не установлены. Одной из причин отсутствия таких данных являются разные подходы к классификации этого порока. Согласно Международной классификации болезней, синдром Арнольда – Киари имеет отдельный шифр (Q07.0), однако определяется в ней как «… патологическое состояние, при котором происходит повышение внутричерепного давления в результате интракраниальной опухоли, окклюзионных форм гидроцефалии, воспалительного процесса, что в некоторых случаях приводит к вклинению мозжечка и продолговатого мозга в большое затылочное отверстие». В ультразвуковой пренатальной литературе до сих пор нам не удалось найти описаний случаев дородовой диагностики синдромаАрнольда- Киари, полностью соответствующих этим характеристикам.
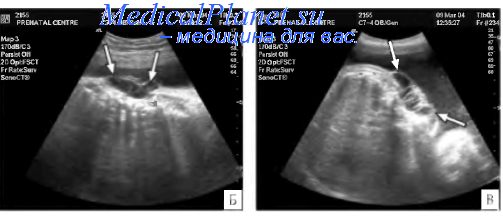
Морфологические особенности различных типов порока Арольда – Киари определяют возможности пренатального выявления и прогноз для жизни. I тип порока совместим с жизнью и нередко является случайной находкой у взрослых при проведении МРТ. Пороки II и III типа в перинатальном периоде встречаются достаточно часто и имеют, как правило, крайне неблагоприятные перинатальные исходы.
Среди диагностированных случаев синдрома Арнольда – Киари основная часть приходится на II тип. Так, в исследованиях СМ. Воеводина из 55 случаев пренатальной диагностики мальформации Арнольда – Киари, которые были зарегистрированы в разные сроки беременности в интервале от 15 до 40 нед I тип составил только 3,5%, II тип – 94,5%, III тип – 2%. По данным нашего центра, при анализе 29 случаев пренатального вывления изменений, которые были нами трактованы, как синдром Арнольда – Киари, лишь у одного плода была обнаружена грыжа в затылочной области, что дает возможность ретроспективно предположить наличие порока III типа (3,4%).
Причины возникновения синдрома Арнольда- Киари до конца не установлены. Хромосомные аномалии при этой патологии, как правило, выявить не удается.
Ультразвуковая диагностика синдрома Арнольда – Киари основана на оценке анатомии структур, расположенных в задней черепной ямке. В норме с конца I триместра при эхографии хорошо видны полушария и червь мозжечка, большая цистерна. В случаях изменения формы и размеров мозжечка, его нечеткой визуализации, уменьшения или исчезновения большой цистерны у врача должно возникнуть подозрение на наличие у плода синдрома Арнольда – Киари.
Очевидно, что пренатальной диагностике в основном доступны II и III тип мальформации Арнольда – Киари. I тип порока может быть заподозрен в тех случаях, когда при ультразвуковом исследовании головы плода в сагиттальной плоскости большая цистерна резко уменьшена в размерах или не визуализируется вовсе. Изображение мозжечка при этом не меняется. Основной морфологический признак этого синдрома – удлинение ствола мозга – по нашему мнению, не имеет четкихэхографическиххарактеристик и поэтому является весьма субъективным.
При II типе порока помимо отсутствия изображения большой цистерны в горизонтальной и сагиттальной плоскостях, отмечается изменение формы и положения мозжечка за счет его каудального смещения. Аномальная форма мозжечка и отсутствие привычного ультразвукового изображения большой цистерны хорошо заметны при эхографии и не вызывают трудностей у врача проводящего исследование.
При III типе порока ко всем описанным выше признакам присоединяется образование затылочной грыжи.
Существенную помощь в пренатальной диагностике синдрома Арнольда- Киари оказывает обнаружение дополнительных эхографических отклонений. В подавляющем большинстве случаев этот порок сопровождается вентрикуломегалией или гидроцефалией, которая является вторичной и легко диагностируется при ультразвуковом исследовании. Поданным отечественных авторов, увеличение ширины тел боковых желудочков мозга в горизонтальной плоскости исследования было обнаружено в 74% случаев порока Арнольда – Киари. Частота этого признака нарастала с увеличением срока беременности (61-85%). Поданным зарубежных авторов, расширение желудочков более 15 мм встречается в 53% случаев синдрома Арнольда-Киари. В наших исследованиях при синдроме Арнольда – Киари у плода изменения желудочковой системы были зарегистрированы в 89,6%.
При эхографическом исследовании плода с подозрением на синдром Арольда – Киари следует обращать внимание и на форму боковых желудочков. Иногдажелудочки меняют ее и становятся заостренными кзади, как бы «ланцетоподобными».
В некоторых случаях синдром Арнольда – Киари сопровождается изменением формы головы («лимон») часто в сочетании с аномальной формой мозжечка («банан»), однако, по мнению СМ. Воеводина, информативность этих признаков низкая. Частота встречаемости этого сочетания в ранние сроки, по его данным, составила около 45%, в интервале 22-28 нед- 24%. Аналогичные данные были приведены М. Van den Hof и соавт. В ранние сроки признак «лимон» был зарегистрирован в 98% случаев, а после 24 нед – только в 13% наблюдений. В наших исследованиях форма головы плода в виде лимона былазарегистрированатолькоу 27,6% плодов с синдромом Арнольда – Киари, при этом во всех случаях срок диагностики не превышал 24 нед.
Помимо горизонтальной и сагиттальной плоскостей в пренатальной диагностике синдрома Арнольда-Киари помощь оказывают и другие плоскости сканирования. Во фронтальной плоскости может отмечаться изменение формы боковых желудочков (70%), атипичность субарахноидальных пространств лобных долей (63%), асимметрия расположения сосудистых сплетений боковыхжелудочков (71 %). Справедливости ради следует отметить, что в практическом здравоохранении в основном используется горизонтальная и реже сагиттальная плоскости, поэтому основное внимание следует обращать на особенности визуализации структур мозжечка и размеры большой цистерны.
Очень важной особенностью синдрома Арнольда-Киари является его сочетание с аномалиями развития позвоночного столба. С одной стороны, в подавляющем большинстве случаев (до 95%) этот комплекс изменений сопровождается образованием спинномозговой грыжи. С другой стороны, патологоанатомические исследования свидетельствуют, что у детей со спинномозговой грыжей частота аномалии развития продолговатого мозга, то есть синдром Арнольда-Киари, приближается к 100%. В наших исследованиях изменения в позвоночнике мы нашли у всех плодов с подозрением на синдром Арнольда – Киари. По мнению некоторых исследователей, именно нарушения в формировании головного мозга, являются основной причиной неудачных исходов лечения спинномозговых грыж у детей.
Наличие или отсутствие спинномозговой грыжи во многом определяет прогноз при синдроме Арнольда – Киари. Учитывая высокую частоту сочетания этих двух пороков и выраженность вторичных изменений в головном мозге (вентрикуломегапия или гидроцефалия) прогноз для жизни и особенно прогноз для постнатального здоровья у плодов с наличием синдрома Арнольда – Киари можно расценивать как неблагоприятный.
Завершая раздел, посвященный вопросам пренатальной диагностики синдрома Арнольда – Киари, следует остановиться на методических особенностях, помогающих поставить точный пренатальный диагноз. Этот синдром по сути своей является пороком развития продолговатого мозга. Аномалии развития позвоночного столба часто сопровождают его, но не являются обязательной составной частью синдрома. Следовательно, пренатальный диагноз синдрома Арнольда – Киари можно ставить только в тех случаях, когда имеются явные изменения в области задней черепной ямки, при этом наличие или отсутствие нарушений строения позвоночника и изменений в желудочковой системе не являются решающими для постановки этого диагноза. Ретроспективный анализ трехлетнего периода, в течение которого были зарегистрированы 29 случаев пренатального выявления изменений у плода, определенных как синдром Арнольда – Киари, показал что в 6 из них ультразвуковые изменения мозжечка не были описаны. В 5 из 6 случаев постнатальные и/или патологоанатомические исследования сняли диагноз синдрома Арнольда – Киари. Как уже указывалось выше, наличие патологии продолговатого мозга существенно ухудшает прогноз для жизни и здоровья, поэтому точный пренатальный диагноз синдрома Арнольда – Киари играет принципиальную роль в определении тактики ведения беременности.
Учебное видео УЗИ головного мозга плода в норме
– Читать далее “Гидроцефалия у плода. Диагностика вентрикуломегалии у плода.”
Оглавление темы “Диагностика патологии нервной системы у плода.”:
1. Методика исследования головного мозга плода. Обследование позвоночника плода.
2. Анэнцефалия плода. Экзэнцефалия плода. Акрания плода.
3. Иниэнцефалия плода. Ранняя диагностика иниэнцефалии плода.
4. Черепно-мозговая грыжа плода. Цефалоцеле у плода.
5. Патология мозолистого тела плода. Обследование мозолистого тела плода.
6. Шизэнцефалия плода. Голопрозэнцефалия у плода.
7. Микроцефалия. Диагностика микроцефалии у плода.
8. Лиссэнцефалия. Диагностика лиссэнцефалии у плода.
9. Как влияют негативные мысли и стресс на развитие плода при беременности?
10. Синдром Арнольда – Киари. Диагностика синдрома Арнольда – Киари у плода.
11. Гидроцефалия у плода. Диагностика вентрикуломегалии у плода.
Источник
Арнольд-Киари – это врожденная патология или аномалия, которая происходит еще в период формирования ребенка внутри утробы матери. Аномалия происходит по причине сдавливания головного мозга, из-за этого деформируются черепные отделы. Последствия таковы: мозжечок и мозговой ствол сильно смещаются и опускаются в затылочную часть, нарушается работоспособность мозга.
Основные причины возникновения аномалии
Хотя медицина быстро развивается, но до сих пор не конца понятно, от чего происходит синдром Арнольда-Киари. Было выявлено, что причина может быть как приобретенного характера, так и врожденная.
Головной мозг
Причина приобретенной болезни:
- Если во время родов произошла черепно-мозговая травма.
- Ликворная травма спинного отдела мозга, из-за чего впоследствии происходит растяжение центрального канала.
Причина врожденной патологии:
- Нехватка питательных веществ для развития плода.
- Неправильное внутриутробное формирование затылочного отверстия, а именно его чрезмерное увеличение.
- Любые дефекты головного мозга.
В любом случае, на протяжении всей беременности нужно наблюдаться у гинеколога. Конечно, на врожденную патологию повлиять невозможно, однако приобретенных патологий можно избежать, если придерживаться всех рекомендаций врача.
Патогенез
Кроме этого, заболевание может развиться:
- Если мама болела венерическими и инфекционными заболеваниями.
- Неконтролируемое количество приемов различных медикаментов в период беременности.
- Употребление табака, наркотических веществ, спиртных напитков.
Конечно, это только предположение, точного показателя нет. Но в любом случае, если беременная женщина не будет следить за своим питанием и вести неправильный образ жизни, это обязательно негативно отразится на развитии ее плода.
Классификация патологии
Синдром Арнольда-Киари делится на подтипы. Общее во всех типах заболевания – это патология низкого прилежания миндалин мозжечка к затылочной части. Отличие – структура черепа, мозга и уровень развития.
Заболевание делится на степени:
- 1 степень включает в себя расположение мозжечковых миндалин в шейном отделе, при этом каких-либо отклонений в развитии головного мозга не происходит
- 2 степень является врожденной патологией, в затылочную часть смещается мозжечок и происходит сдавливание спинного мозга, позвоночника и головы. Обнаружить аномалию данного типа можно только при инструментальном обследовании
- 3 тип обусловлен наличием мозга продолговатой формы, так называемая грыжа, мозжечок опускается ниже нормы, последствия могут быть критическими
- 4 степень – полное нарушение функциональности мозжечка, при такой ситуации плод не выживает
Симптоматика болезни
Симптомы имеют как индивидуальный, так и общий характер. Также все зависит от возраста больного, иногда данное заболевание может появиться в зрелом возрасте. Кроме того, симптоматика отличается в зависимости от подтипа заболевания.
Болезнь Арнольда-Киари первой степени. Симптомы проявляются очень выраженно у взрослых больных:
Головная боль
- перманентная головная боль
- парестезия, сильная слабость в руках
- тошнота
- диспепсия
- диплопия
- покачивание при ходьбе
- боль и шум в ушах
- остеохондроз шейного позвонка
- нечеткая речь
- проблемы с глотанием
Признаки 2-го типа патологии Арнольда-Киари наблюдаются уже после рождения малыша, более выражены во время грудного вскармливания:
- нарушение глотательной функции
- негромкий и слабый крик малыша
- нарушение функциональности дыхательных путей: слышны шумы и свист
Третья и четвертая степень болезни протекают очень тяжело, чаще всего происходит смерть пациента из-за инфаркта спинного мозга. Характерны следующие признаки:
- расстройство координации
- наличие тремора
- диплопия или нечеткая видимость в момент вращения головы
- обморочное состояние
- головокружение
- обширная потеря чувствительности
- задержка мочеиспускания
- слабость в мышцах конечностей и лица
Лечение назначит врач только после точной диагностики и степени развития заболевания.
Диагностика болезни
Бывают случаи, когда данная аномалия никак не проявляется, полностью отсутствуют симптомы. Тогда про болезнь можно узнать при помощи специального медицинского обследования и даже при обычном осмотре врача.
Диагностика болезни
Если у врача возникает подозрение на болезнь Арнольда-Киари, то он назначает такие процедуры: электроэнцефалограмма головного мозга, эхоэнцефалография (ЭХО-ЭГ) и реоэнцефалография. Все эти исследования позволяют измерить внутричерепное давление, и увидеть даже самые незначительные патологии. Однако окончательный диагноз по таким снимкам не ставится.
Проводится дополнительное обследование. Пациента направляют на резонансно-магнитную томографию или компьютерную томографию. Желательно во время проведения такой процедуры оставаться неподвижным, чтобы получить изображение, приближенное к оригиналу. Так как маленьких детей нельзя заставить не двигаться пару минут, то им в кровь вводится препарат, вводящий малыша в медикаментозный сон. Томография помогает обнаружить дефект костной структуры и выявит различные образования в черепе больного.
Также для установки диагноза назначают томографию позвоночника, которая показывает, нет ли еще каких-либо патологий. Беременным назначают повторное УЗИ.
Все процедуры следует проводить четко по рекомендации врача. Настоящий специалист не будет ставить точный диагноз, не проведя должного обследования.
Эффективное лечение синдрома
На сегодняшний момент используют два вида лечения: хирургическое, когда дело доходит до операции, и консервативное.
Лечение
Консервативное лечение используют в том случае, когда болезнь не причиняет больному сильного дискомфорта и не отражается на его развитии. Врач рекомендует чаще заниматься физкультурой, упражнениями для мышечной координации. Также прописываются некоторые препараты: обезболивающее, для расслабления мышц, противовоспалительное средство. Дополнительно назначается комплекс витаминов, особенно группы B, так как они отвечают в организме за биохимические процессы и нормализуют работу центральной нервной системы.
Конечно, такие назначения не помогут полностью избавиться от болезни, но позволят максимально долго обходиться без хирургического вмешательства.
Если мальформация болезни прогрессирующая, тогда понадобится неотложное хирургическое вмешательство. Делается или операция, или шунтирование. Операция позволяет решить две основных причины:
- Исправить дефекты, которые способствуют сдавливанию черепа и головного мозга.
- Приводит в нормальное состояние движение ликвора.
Такая операция довольно распространенная, длительность ее не более двух часов. Пациент восстанавливается полностью через пару недель. Благодаря операции внутричерепное давление нормализуется, а пространство в спинном и головном мозге увеличивается, болезнь отступает.
Профилактические меры
Всегда нужно бережно относиться к своему здоровью, а если это период, когда женщина вынашивает под сердцем ребенка, тогда ответственность удваивается. Существуют некоторые профилактически меры, позволяющие предотвратить заболевание:
- включить в свой рацион больше фруктов и овощей
- пить свежие соки, кушать молочные изделия и мясо, богатые белком
- пить витамины для беременных
- отказаться от вредных привычек, если таковые имеются
- пить только те препараты, которые допустимы во время беременности и только по назначению врача
- проводить все необходимые обследования
Если следить за своим питанием и вести полноценный, здоровый образ жизни, вовремя сдавать анализы и прислушиваться к врачу, то вероятность родить здорового ребенка увеличивается во много раз.
Итак, синдром Арнольда-Киари у плода возникает по разным причинам, как врожденным, так и приобретенным. Заболевание 1-го и 2-го типа полностью излечимо, если провести необходимую операцию. Чтобы предотвратить появление патологии, будущей маме необходимо максимально позаботиться о своем здоровье, тем самым это будет положительно сказываться на развитии головного мозга ее плода.
Заметили ошибку? Выделите ее и нажмите Ctrl+Enter, чтобы сообщить нам.
Фев 25, 2017Виолетта Лекарь
Источник
Синдром Арнольда-Киари — врожденная патология краниовертебральной зоны, возникающая в период эмбриогенеза и характеризующаяся деформацией черепа. Опущение структур головного мозга в большое затылочное отверстие приводит к их сдавлению и ущемлению. Дисфункция мозжечка сопровождается нистагмом, шаткой походкой, дискоординацией движений, а поражение продолговатого мозга — изменениями в работе жизненно важных органов и систем.
Впервые синдром был описан в конце 18 века двумя учеными: доктором из Германии Арнольдом Джулиусом и врачом из Австрии Гансом Киари. Аномалию обнаруживают сразу после появления ребенка на свет или несколько позже — в пубертатном или зрелом возрасте. Это зависит от типа синдрома. У взрослых пациентов недуг чаще всего становится неожиданной находкой. Средний возраст больных — 25-40 лет.
Симптоматика патологии также определяется типом мальформации. Хоть синдром и считается врожденным пороком, не всегда его клинические признаки возникают с самого рождения. Иногда они обнаруживаются после 40 лет. У больных возникает цефалгия, напряженность мышц шеи, головокружение, нистагм, обмороки, атаксия, нарушения речи, парез гортани, тугоухость, падение остроты зрения, нарушение процесса глотания, стридор, парестезии, слабость мышц. У большинства пациентов симптомы заболевания полностью отсутствуют. Его обнаруживают случайно, во время комплексного диагностического обследования, проводимого совсем по другому поводу. Пациентам с бессимптомными формами не требуется лечение.
При постановке диагноза учитывают данные осмотра больного и его неврологического статуса, а также результаты томографического исследования мозга. Магнитно-ядерный резонанс позволяет точно и быстро определить наличие трансформации, уровень поражения и степень ущемления головного мозга. Лечение синдрома медикаментозное, физиотерапевтическое и оперативное. Больным выполняют операции по шунтированию мозга и декомпрессии краниовертебральной зоны.
Этиология
Причины синдрома в настоящее время точно не определены. Существует несколько теорий происхождения патологии, но до конца ни одна из них не имеет официального подтверждения. Мнения ученых-неврологов всего мира до сих пор расходятся.
Большинство медиков признают синдром врожденным недугом, сформированным в процессе эмбриогенеза под воздействием негативных факторов внешней среды, оказывающих свое пагубное действие на женский организм при беременности. К ним относятся: самостоятельные использование лекарств, алкоголизм, табакокурение, вирусные инфекции, ионизирующее излучение.

Врожденные причины:
- Дистопия мозжечковых миндалин за пределы задней черепной ямки, имеющей относительно малые размеры;
- Выталкивание растущих структур мозга через затылочное отверстие;
- Неправильное формирование в процессе эмбриогенеза и атипичное развитие в постнатальном периоде костной ткани, приводящее к деформации черепной коробки.
Некоторые ученые определенную роль в развитии синдрома отводят генетическому фактору. В настоящее время можно точно утверждать, что болезнь не связана с хромосомными аномалиями.
Другие ученые придерживаются иной точки зрения относительно происхождения синдрома. Они считают его приобретенным и объясняют свое мнение появлением симптомов патологии у взрослых лиц. Приобретенный синдром возникает под действием экзогенных факторов. У больного новорожденного ребенка череп может иметь нормальное строение без костных аномалий и гипоплазии.
Приобретенные причины:
- Родовой травматизм с поражением черепа и мозга,
- Воздействие цереброспинальной жидкости на стенки спинного мозга,
- Любые ЧМТ,
- Бурный рост мозга в условиях медленно растущих костей черепа.
Симптомы патологии долгое время отсутствуют у больных, а затем внезапно появляются под воздействием провоцирующих факторов: вирусов, травм головы, стрессов.
Опущение основных мозговых структур до шейных позвонков блокирует процесс перетекания ликвора из подпаутинного пространства в спинномозговой канал. Это приводит к дисциркуляторным изменениям. Ликвор, продолжая синтезироваться и никуда не оттекая, скапливается в головном мозге.
Ученый Киари в 1891 году выделил четыре типа аномалии:
- I – выход структурных элементов головного мозга за пределы задней черепной ямки, обусловленный недоразвитием костной ткани этой области. Этот тип клинически проявляется у лиц зрелого возраста.

выход структур мозжечка за пределы ЗЧЯ при аномалия 1 типа
- II — нарушения эмбриогенеза, приводящие к расположению структур мозжечка и продолговатого мозга ниже большого затылочного отверстия.

синдром Арнольда-Киари II типа
- III — эктопия мозговых структур в каудальном направлении с образованием энцефаломенингоцеле.

аномалия III типа
- IV — недоразвитый мозжечок не смещается и не выходит за пределы черепа. Поскольку отсутствует грыжевое выпячивание мозга, этот тип синдрома отсутствует в современной классификации.
Существуют два новых типа синдрома. Тип — мозжечок располагается достаточно низко, но при этом находится в черепной коробке. Тип 1.5 – промежуточная форма, сочетающая признаки I и II типов.
Выделяют три степени тяжести патологии:
- Первая — относительно легкая форма патологии без аномалий мозговых структур и характерных клинических проявлений.
- Вторая — наличие пороков развития ЦНС с врожденным недоразвитием головного мозга и подкорки.
- Третья — аномалии строения головного мозга со смещением мягких тканей в отношении твердых структур, образованием ликворных кист и сглаженностью извилин.
Симптоматика

Аномалия Киари I типа — самая распространенная форма синдрома, клинические признаки которой условно объединены в пять синдромов:
- Гипертензионный синдром проявляется цефалгией, подъемом артериального давления в утренние часы, напряженностью и гипертонусом шейных мышц, дискомфортом и болезненными ощущениями в шейном отделе позвоночника, диспепсическими явлениями, общей астенизацией организма У новорожденных детей возникает общее беспокойство, рвота фонтаном, тремор подбородка и конечностей, нарушается сон. Ребенок постоянно плачет, отказывается от груди.
- При наличии мозжечковых нарушений у больных изменяется произношение, речь становится скандированной, возникает вертикальный нистагм. Они жалуются на частые головокружения, рассогласованность движений, шаткость походки, дрожание рук, нарушение равновесия, дезориентацию в пространстве. Больные с большим трудом выполняют простые целенаправленные действия, в движениях отсутствует четкость и скоординированность.
- Поражение черепно-мозговых нервов проявляется признаками корешкового синдрома. У пациентов ограничивается подвижность языка и мягкого неба, что приводит к нарушению речи и проглатывания пищи. Их голос изменяется в сторону гнусавости и осиплости, речь становится неясной, дыхание затрудненным. Нарушение ночного дыхания отмечаются у большинства больных. У них возникает гипопноэ, центральное или обструктивное апное, при прогрессировании которого развивается острая дыхательная недостаточность. Лица с синдромом плохо слышат и видят, у них двоится в глазах и шумит в ушах. Со стороны органов зрения пациенты отмечают наличие светобоязни и боль при движении глазными яблоками. Офтальмологи часто обнаруживают анизокорию, спазм аккомодации или скотомы. Одним из основных симптомов синдрома является гипестезия – снижение чувствительности кожи лица и конечностей. Подобные патологическое изменения связаны с приглушенным реагированием рецепторов кожи на внешние раздражители: тепло или холод, уколы, удары. В тяжелых случаях нервное окончания вообще перестают воспринимать различные экзогенные воздействия.
- Сирингомиелический синдром — сложный симптомокомплекс, проявляющийся парестезией или онемением конечностей; изменением тонуса мышц и их гипотрофией, приводящей к миастеническим расстройствам; поражением периферических нервов, проявляющимся болью в конечностях; дисфункцией органов таза в виде затрудненной дефекации или самопроизвольного мочеиспускания; возможны артропатии — поражения суставов.
- У больных с пирамидальной недостаточностью снижается сила в нижних конечностях и способность к тонким движениям, ограничивается объем движений, повышается мышечный тонус — так называемая спастичность, например, спастическая походка. Повышение сухожильных рефлексов сочетается с одновременным снижением кожных рефлексов — брюшных. Возможно появление патологических рефлексов. У пациентов страдает мелкая моторика рук.
Любое неосторожное движение усиливает симптомы патологии, делает их более выраженными и яркими. Изменение положения головы — частая причина потери сознания.
Синдром Киари II типа имеет сходные клинические проявления. У новорожденных возникает паралич гортани, врожденный стридор, ночное апноэ, дисфагия, срыгивания, нистагм, гипертонус мышц рук, цианоз кожи. Аномалии III и IV типов не совместимы с жизнью.
Диагностические мероприятия

Аномалия Арнольда-Киари на снимке МРТ
Врачи-неврологи и невропатологи осматривают пациента и выявляют характерные особенности походки, изменение рефлексов и чувствительности на определенных участках тела, слабость в руках и прочие признаки. Все проявления мозжечкового, гидроцефального, бульбарного и прочих синдромов в совокупности позволяют врачу заподозрить аномалию.
После определения неврологического статуса больного требуется проведение комплексного неврологического обследования, включающего инструментальные методы — электроэнцефалографию, УЗИ головного мозга, реоэнцефалографию, ангиографию, рентгенографию. Эти методики выявляют лишь косвенные признаки патологии — изменения, происходящие в организме больного.
Ядерный магнитный резонанс лежит в основе особого нерентгенологического метода исследования – томографии. Этот спектроскопический анализ безопасен для большинства людей. Он дает изображение, состоящее из тонких срезов от магнитнорезонансного сигнала, проходящего через тело человека. На сегодняшний день именно МРТ позволяет быстро и точно поставить диагноз. Томография визуализирует структуру костей и мягкие ткани черепа, определяет пороки мозга и его сосудов.
Лечебный процесс
Лечение аномалии Киари комплексное, включающее медикаментозное воздействие, физиотерапевтические процедуры и хирургическое вмешательство. Именно оно в большинстве случаев помогает справиться с недугом и восстановить нормальную работу всего организма. Возможно применение средств народной медицины, которые дополняют, но не заменяют основное лечение. Использование фитосборов, отваров и настоев лекарственных трав должно быть одобрено лечащим врачом.
Лекарственная терапия и физиотерапия
Если больные испытывают сильную головную боль, боль в шее, мышцах и суставах, им назначают следующие группы препаратов:
- Обезболивающие средства – «Кеторол», «Пенталгин», «Анальгин».
- НПВС для уменьшения боли – «Мелоксикам», «Ибупрофен», «Вольтарен».
- Миорелаксанты для снятия напряжения с мышц шеи – «Мидокалм», «Сирдалуд».
Патогенетическое лечение синдрома включает:

- Препараты, улучшающие мозговое кровообращение – «Пирацетам», «Винпоцетин», «Циннаризин».
- Диуретики для уменьшения образования ликвора и с целью дегидратации – «Фуросемид», «Маннитол».
- Витамины группы В, поддерживающие работу нервной системы на оптимальном уровне и оказывающие антиоксидантное действие – «Тиамин», «Пиридоксин». Наиболее распространенные витаминные средства «Мильгамма», «Нейромультивит», «Комбилипен».
Если состояние больного признают крайне тяжелым, то его госпитализируют сразу в реанимационное отделение. Там пациента подключают к аппарату ИВЛ, устраняют имеющийся отек мозга, предупреждают инфекционные патологии и корректируют неврологические нарушения.
Физиотерапевтическое воздействие дополняет медикаментозное лечение, позволяет быстрее добиться положительных результатов, ускоряет процессы восстановления функций организма и выздоровления больных. Неврологи назначают:
- Криотерапию, оказывающую обезболивающий эффект, стимулирующую работоспособность желез внутренней секреции и укрепляющую иммунитет.
- Лечение лазером, улучшающее трофику и микроциркуляцию в очаге поражения.
- Магнитотерапию, оказывающую общее оздоравливающее действие и запускающую внутренние резервы организма.
В настоящее время особой популярностью пользуется кинезиологическая терапия, которая направлена на развитие умственных способностей и достижение физического здоровья через двигательные упражнения. Ее также включают в схему лечения данного синдрома.
Лечение не проводится вообще, если патология была обнаружена случайно, во время прохождения томографического обследования совсем по другому поводу, и у больного отсутствуют какие-либо характерные симптомы. За состоянием таких пациентов специалисты ведут динамическое наблюдение.
Оперативное вмешательство
Стойкие неврологические нарушения с парестезиями, дистонией мышц, параличами и парезами требуют проведения хирургической коррекции. Оперативное вмешательство показано также в тех случаях, когда медикаментозная терапия на дает положительного результата. Операции преследуют одну цель — устранение сдавления и ущемления мозга, а также восстановление нормальной циркуляции ликвора.

В настоящее время нейрохирурги спасают жизнь больным путем выполнения декомпрессивных и шунтирующих операций. В первом случае выпиливают часть затылочной кости с целью расширения большого отверстия, а во втором создают обходной путь для оттока ликвора по имплантационным трубкам с целью снижения его объема и нормализации внутричерепного давления.
После хирургического вмешательства всем пациентам показаны реабилитационные мероприятия. Если лечение было успешным, у больных восстанавливаются утраченные функции — дыхательные, двигательные, сердечно-сосудистые, нервные. В течение трех лет возможно рецидивирование патологии. В таких случаях больных признают инвалидами.
Видео: об операции при синдроме Арнольда-Киари
Народная медицина
Народные средства, применяемые при данной патологии, устраняют боль и расслабляют напряженные мышцы. Они эффективно дополняют традиционную терапию синдрома.
Наиболее популярные средства:
- Настой алтея для постановки компрессов,
- Прогревания пор?