
Наследственные синдромы сочетающиеся с сахарным диабетом

Статья из монографии “Сахарный диабет: от ребёнка до взрослого”.
Синдром Дауна – трисомия по 21 паре хромосом. Внешний вид больного ребенка: плоское лицо, монголоидный разрез глаз, эпикант, открытый рот, короткий нос, плоская переносица, страбизм, брахицефалия, плоский затылок, диспластические уши, зубные аномалии, короткая шея, брахимезофалангия, клинодактилия 5–го пальца кисти, мышечная гипотония, поперечная ладонная складка, умственная отсталость.
Синдром Кляйнфельтера – полисомия по Х–хромосоме у лиц мужского пола. Данный синдром проявляется у мужчин и мальчиков–подростков в виде евнухоидных пропорций тела, гинекомастии, недоразвития гонад, иногда – ожирения. Данная генетическая патология является самой распространенной причиной мужского гипогонадизма и бесплодия.
Синдром Тернера – аномалия или отсутствие одной Х–хромосомы у лиц женского пола. В периоде новорожденности отмечается лимфатический отек конечностей, диагностируют пороки сердца. Пороки развития скелета и внутренних органов обнаруживают в более старшем возрасте. Для данной генетической патологии характерны низкорослость, дисгенезия или полное отсутствие гонад, короткая шея с крыловидными кожными складками, низкая линия роста волос на затылке, бочкообразная грудная клетка, О–образное искривление рук, Х–образное искривление ног.
Синдром Вольфрама, или синдром DIDMOAD (Diabetes Insipidus – диабет, Diabetes Mellitus – сахарный диабет, Optic Atrophy – атрофия зрительного нерва, Deafness – глухота), – прогрессирующее нейродегенеративное заболевание с аутосомно–рецессивным типом наследования и манифестацией до 16–летнего возраста. Особенно часто встречается среди народностей, у которых отмечаются близкородственные браки.
Сахарный диабет (СД) и атрофия зрительного нерва развиваются в первую декаду жизни, во вторую декаду – нейросенсорная тугоухость и несахарный диабет. В третьей и четвертой декадах жизни нередко развиваются другие клинические проявления: психические изменения, атония мочевыводящих путей и дилатация мочевыводящего тракта, атаксия. Мутация гена WFS1 синдрома Вольфрама, кодирующего полипептид вольфрамин, обнаруживается у 90% пациентов с клиническими проявлениями данного синдрома.
Синдром Роджерса – сочетание сахарного диабета, мегалобластной анемии, рефрактерной к витамину В12 и фолиевой кислоте, нейросенсорной глухоты. В патогенезе имеет место нарушение обмена тиамина (витамина В1). Данный синдром является результатом мутации гена SLC19A2. Сахарный диабет развивается вследствие инсулиновой недостаточности, иногда чувствителен к назначению тиамина, но в дальнейшем все же требуется назначение инсулина. Глухота при назначении тиамина не имеет своего обратного развития.
Атаксия Фридрейха – редкое наследственное заболевание, характеризующееся потерей произвольных мышечных движений (атаксией) и кардиомегалией. Данная патология встречается в равной степени у мальчиков и девочек. Заболевание имеет аутосомно–рецессивный тип наследования и связано с поломкой гена фратаксина, локализованного на 9 хромосоме. Фратаксин находится в митохондриях, но до настоящего времени окончательно функция его не установлена.
Хорея Геттингтона – нейродегенеративное наследственное заболевание, приводящее к деменции. Ген, ответственный за данное заболевание, локализован на 4 хромосоме и представляет собой триплет, кодирующий синтез протеина – хунтингтина.
Синдром Лоренса–Муна–Бидля (рис. 4.3). Классическими клиническими проявлениями синдрома являются задержка умственного развития, пигментный ретинит, гипогонадизм, аномалии развития глаз, почек. Иногда имеет место полидактилия. Для заболевания характерен аутосомно–рецессивный тип наследования. У мальчиков данный синдром встречается в 2 раза чаще. Основные симптомы заболевания проявляются в первые 2–3 года жизни. Зрительные нарушения могут приводить к слепоте уже к 8 годам. По данным магнитно–резонансной томографии (МРТ) головного мозга у больных с синдромом Лоренса–Муна–Бидля наблюдается симптом «пустого» турецкого седла.
Рис. 4.3. Синдром Лоренса-Муна-Бидля
Миотоническая дистрофия относится к группе наследственных нервно–мышечных заболеваний вследствие нарушений медиаторного обмена. Болезнь начинается с затруднения расслабления мышц, с последующим нарастанием мышечной атрофии (слабости), выпадением сухожильных рефлексов, исхуданием, кахексией или ожирением. Очень часто наблюдается бесплодие, инфантилизм. Имеет место выпадение волос, катаракта, умеренная психическая недостаточность. Болезнь может манифестировать в различном возрасте, но чаще – в раннем пубертатном. Причина данного заболевания – патология гена, локализованного в 19 хромосоме и кодирующего фермент киназу скелетной мускулатуры.
Порфирия (пурпурный, греч.) – группа заболеваний, связанная с патологией синтеза гема. Пурпурная окраска мочи, характерная для данной патологии, послужила поводом для использования этого термина. Гем является не только центральной кислородсвязывающей частью гемоглобина, но и активной частью жизненно важных энзимов цитохромов (Р 450 и его предшественников). Гены, ответственные за синтез энзимов, расположены в различных хромосомах и при их мутациях патология имеет различный (аутосомно–рецессивный или аутосомно–доминантный) тип наследования.
Носители патологического гена, как правило, не имеют клинических проявлений. Часто в клинической картине пациентов с порфирией имеются патология ЖКТ, гепатиты, повышенная фотосенсибилизация. Иногда у больных отмечаются судороги, мышечная слабость.
Синдром Прадера–Вилли характеризуется умственной отсталостью, мышечной гипотонией, низкорослостью, эмоциональной лабильностью и нарушением аппетита с формированием ожирения. Данный синдром является результатом отсутствия сегмента 11–13 в длинном плече 15 хромосомы либо его делеции (у 70–80% больных). Клинические проявления зависят от возраста. В раннем возрасте отмечаются низкая двигательная активность и мышечная гипотония. Затем появляется недостаточность психического и физического развития. Значительная прибавка в весе отмечается в возрасте от 1 года до 6 лет. Характерными проявлениями этого синдрома являются маленькие стопы и кисти, миндалевидные глаза, «тентовидная» верхняя губа, гипогонадизм. У пациентов может наблюдаться синдактилия.
Из монографии «Сахарный диабет: от ребенка до взрослого»
Сенаторова А.С., Караченцев Ю.И., Кравчун Н.А., Казаков А.В., Рига Е.А., Макеева Н.И., Чайченко Т.В.
ГУ «Институт проблем эндокринной патологии им. В.Я. Данилевского АМН Украины»
Харьковский национальный медицинский университет
Харьковская медицинская академия последипломного образования МЗ Украины
Источник

Неонатальный сахарный диабет – это группа метаболических патологий, которые проявляются гипергликемией и гипоинсулинемией на фоне дисфункции поджелудочной железы. Клинические симптомы включают в себя гипергликемию, полиурию, метаболический ацидоз, кетонемию и кетонурию, дегидратацию, дефицит массы тела. Другие проявления зависят от основной генетической патологии. Диагностика основывается на физикальном осмотре ребенка, лабораторных анализах крови и мочи. Лечение зависит от формы неонатального сахарного диабета. При транзиторном варианте проводится симптоматическая коррекция гипергликемии, при перманентном диабете показана пожизненная инсулинотерапия.
Общие сведения
Неонатальный сахарный диабет (НСД) – это совокупность гетерогенных патологий в неонатологии и педиатрии, для которых характерна гипергликемия и транзиторная или перманентная недостаточность инсулина на фоне дисфункции β-клеток эндокринной части поджелудочной железы. Впервые сахарный диабет у новорожденного описал Kistel в 1852 году. Распространенность данного состояния составляет 1:300-400 тыс. новорожденных. В 55-60% случаев развивается транзиторная форма. Перманентный НСД встречается реже, и, как правило, входит в состав синдромологических патологий. В среднем мальчики и девочки болеют с одинаковой частотой, однако некоторые синдромы (например, IPEX-синдром) более характерны для мужского пола. Тип наследования определенных форм неонатального сахарного диабета также зависит от конкретной генетической аномалии и может быть как аутосомно-доминантным (дефект GK), так и аутосомно-рецессивным (KCNJ11).

Неонатальный сахарный диабет
Причины неонатального сахарного диабета
Этиология неонатального сахарного диабета зависит от его клинической формы. Преходящий НСД возникает в результате неполноценного развития β-клеток островков Лангерганса поджелудочной железы. Функционально незрелые клетки неспособны обеспечить адекватную реакцию на повышение гликемии. При этом базовый уровень инсулина в плазме крови может быть нормальным. В большинстве случаев патология развивается спорадически. Также доказана наследственная склонность, связанная с аномалиями длинного плеча VI хромосомы. Причиной возникновения транзиторного неонатального сахарного диабета могут быть мутации генов ABCC8 и KCNJ11, однако дефекты этих же генов в ряде случаев провоцируют развитие перманентной формы.
Персистирующий неонатальный сахарный диабет обусловлен аномалиями структуры β-клеток, всей железы или непосредственно инсулина, из-за чего развивается его абсолютная недостаточность. Как правило, это наследственные дефекты различных генов. Наиболее распространенными вариантами являются гетерозиготные активации мутации генов ABCC8 и KCNJ11. Часто встречаются следующие аномалии, обуславливающие развитие НСД: IPF-1 – гипо- или аплазия поджелудочной железы, GK – отсутствие реакции на уровень глюкозы в крови, EIF2FK3 (синдром Уолкотта-Раллисона) – нарушение процесса синтеза инсулина, FOXRЗ (IPEX-синдром) – аутоиммунное поражение тканей железы. Перманентная форма также может быть проявлением митохондриальных патологий. В некоторых случаях спровоцировать развитие неонатального сахарного диабета может энтеровирусная инфекция, которую мать перенесла в первом триместре беременности.
Классификация и симптомы неонатального сахарного диабета
Неонатальный сахарный диабет имеет две основных клинических формы:
- Преходящий или транзиторный НСД. Более распространенный вариант. Независимо от проводимого лечения симптомы постепенно исчезают в возрасте до 3 месяцев. Полная ремиссия наступает в возрасте от 6 месяцев до 1 года. Возможны рецидивы во взрослом возрасте.
- Персистирующий или перманентный НСД. Часто входит в структуру синдромальных пороков развития. Требует пожизненной инсулинотерапии.
Клинические проявления транзиторного и перманентного неонатального сахарного диабета при отсутствии других синдромальных нарушений практически идентичны. При преходящем НСД часто наблюдается задержка внутриутробного развития – дети рождаются с массой тела значительно меньше нормы (ниже 3 перцентиля) для своего срока гестации. Общее состояние ребенка при транзиторной форме нарушено незначительно – пациент малоподвижный, вялый, аппетит снижен или сохранен. Коматозные состояния нехарактерны. Даже на фоне полноценного питания ребенок медленно прибавляет к массе тела. Специфическим признаком неонатального сахарного диабета является выраженная полиурия и дегидратация, часто – резкий запах ацетона изо рта.
Для перманентной формы неонатального сахарного диабета характерны все вышеперечисленные симптомы, но большей интенсивности. Несмотря на это, задержка внутриутробного развития выражена не так сильно. Другие возможные симптомы зависят от того, входит ли НСД в структуру того или иного синдрома. При развитии IPEX-синдрома гипергликемия сочетается другими эндокринными и иммунными нарушениями и целиакнегативной энтеропатией. Клинически это проявляется экземой, хронической диареей, аутоиммунным тиреоидитом, гемолитической анемией. Синдром Уолкотта-Раллисона помимо неонатального сахарного диабета включает почечную недостаточность, нарушения интеллекта, гепатомегалию и спондилоэпифизарную дисплазию.
Диагностика неонатального сахарного диабета
Диагностика неонатального сахарного диабета включает в себя физикальный осмотр новорожденного, лабораторные и инструментальные методы исследования. Сбор анамнестических данных у матери, как правило, позволяет определить выраженность уже имеющихся проявлений – полиурии, медленной прибавки к массе тела. При объективном обследовании выявляется общая адинамия ребенка, сухость кожных покровов и другие проявления дегидратации. В большинстве случаев у детей наблюдается отставание в физическом развитии и дефицит массы тела.
Лабораторные анализы играют ведущую роль в диагностике неонатального сахарного диабета. При исследовании крови определяется стабильная гипергликемия более 10-11 ммоль/л, повышение уровня кетоновых тел свыше 3 ммоль/л, рН<7,3 – метаболический ацидоз. В анализе мочи можно обнаружить глюкозурию и кетонурию. При проведении пробы по Зимницкому выявляется увеличение суточного диуреза (полиурия) и повышение удельного веса мочи. Инструментальная диагностика в виде УЗД, рентгенографии ОБП, ультрасонографии используется для исключения органических нарушений и определения других проявлений синдромальных патологий. При возможности проводится кариотипирование с идентификацией дефектных генов.
Дифференциальный диагноз неонатального сахарного диабета осуществляется с гипергликемией новорожденных, гликозурией на фоне массивной инфузионной терапии, внутриутробными инфекциями, воспалительными заболеваниями ЦНС – менингитом и энцефалитом, почечным диабетом, инфекциями кишечного тракта, синдромом мальабсорбции, острыми хирургическими абдоминальными патологиями.
Лечение неонатального сахарного диабета
Терапевтическая тактика при перманентной и транзиторной формах неонатального сахарного диабета существенно отличается. Детям с персистирующим НСД показана заместительная инсулинотерапия, которая дополняется высококалорийным питанием. Терапевтическая схема подбирается индивидуально для каждого ребенка на основе чувствительности к инсулину и уровня глюкозы в крови. Как правило, применяют инсулины как короткого, так и длительного действия. В зависимости от присутствующей синдромальной патологии неонатального сахарного диабета проводится соответствующая коррекция. Например, при мутации гена FOXRЗ назначаются цитостатики, выполняется пересадка костного мозга, а при дефекте KCNJ11 вместо инсулинов используются препараты сульфанилмочевины. Заместительная инсулинотерапия показана на протяжении всей жизни.
У больных с транзиторной формой неонатального сахарного диабета инсулинотерапия используется только при высоких уровнях гликемии, эксикозе, выраженном нарушении общего состояния, дефиците массы тела и ее медленном наборе. На протяжении первых 6-12 месяцев потребность в сахароснижающих препаратах уменьшается, а затем пропадает – наступает полная ремиссия. Контроль над уровнем глюкозы крови и коррекция доз препаратов в зависимости от динамики НСД может проводиться каждые 7 дней или 1 раз в месяц у эндокринолога и педиатра или семейного врача.
Прогноз и профилактика
Прогноз при транзиторной форме неонатального сахарного диабета благоприятный. Как правило, в возрасте от 6 месяцев до 1 года наступает полная клиническая ремиссия. У части детей в дальнейшем может наблюдаться нарушение толерантности к глюкозе. Также существует риск развития аутоиммунного диабета в возрасте 20-30 лет. Прогноз в отношении выздоровления при перманентной форме неонатального сахарного диабета неблагоприятный. Вне зависимости от присутствующих патологий ребенок будет вынужден пожизненно принимать инсулин. Прогноз для жизни при этой форме НСД сомнительный. Исход во многом зависит от наличия тех или иных генетических нарушений. При IPEX-синдроме большинство детей умирают в возрасте до 1 года от тяжелых форм сепсиса.
Специфической профилактики неонатального сахарного диабета не разработано. Неспецифические превентивные меры включают в себя медико-генетическое консультирование семейных пар с оценкой вероятности рождения ребенка с данной патологией. При высоком риске возникновения НСД у будущего ребенка возможно проведение амниоцентеза с последующим кариотипированием.
Источник
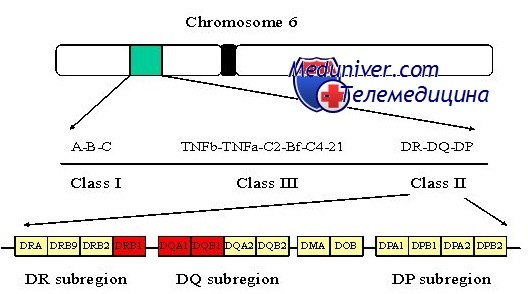
Генетика сахарного диабета I типа. Особенности наследованияСуществуют два основных типа сахарного диабета: I тип (инсулинзависимый — ИЗСД) и II тип (инсулиннезависимый — ИНСД), составляющие 10 и 88% всех случаев соответственно. Они отличаются типичным возрастом начала, конкордантностью однояйцовых близнецов и ассоциацией с конкретными аллелями главного комплекса гистосовместимости (МНС — major histocompatibility complex). Семейное накопление наблюдают при обоих типах сахарного диабета, но в одной семье обычно присутствует только I или II тип. Сахарный диабет I типа встречается в белой популяции с частотой около 1 на 500 (0,2%), в африканских и азиатских популяциях — реже. Обычно его обнаруживают в детстве или юности, и он вызван аутоиммунным поражением b-клеток поджелудочной железы, вырабатывающих инсулин. У преобладающего большинства больных детей уже в раннем детстве, задолго до развития явных проявлений болезни, вырабатываются многочисленные аутоантитела против ряда эндогенных белков, включая инсулин. Ассоциация главного комплекса гистосовместимости при сахарном диабете I типаПри I типе сахарного диабета существует подтверждение роли генетических факторов: конкордантность однояйцовых близнецов приблизительно 40%, что далеко превышает 5% конкордантности у разнояйцовых. Риск диабета I типа для сибсов больного пробанда около 7%, что дает показатель наследуемости hs = 7% / 0,2% =- 35. Давно известно, что локус МНС — основной генетический фактор при сахарном диабете, так как около 95% всех пациентов с сахарным диабетом I типа (по сравнению с примерно 50% в нормальной популяции) — гетерозиготные носители аллелей HLA-DR3 или HLA-DR4 в локусе HLA класса II в МНС [HLA — человеческие лейкоцитарные антигены (human leucocyte antigens)]. Первое исследование, показавшее ассоциацию HLA-DR3 и HLA-DR4 с сахарным диабетом I типа при использовании стандартных методов проверки достоверности различия между разными аллелями HLA, проводили методом иммунологических реакций in vitro. Позже этот метод заменили прямым определением ДНК-последовательности разных аллелей. Секвенирование локуса гистосовместимости у огромного количества больных обнаружило, что «аллели» DR3 и DR4 — не просто аллели.
Как DR3, так и DR4 могут быть подразделены на десятки аллелей, располагающихся в локусе, теперь называющемся DRB1, и определяемых на уровне последовательности ДНК. Кроме того, стало ясным, что ассоциация между определенными аллелями DRB1 и сахарным диабетом I типа частично вызвана аллелем в другом локусе класса II, DQB1, располагающимся примерно в 80 килобазах от DRB1, вместе формирующих общий гаплотип (вследствие неравновесного сцепления; см. главу 10) друг с другом. DQB1 кодирует b-цепь, одну из цепей, формирующих димер белка класса II DQ. Оказывается, что присутствие аспарагиновой кислоты (Asp) в 57 позиции b-цепи DQ тесно связано с устойчивостью к сахарному диабету I типа, тогда как другие аминокислоты в этом положении (аланин, валин или серии) определяют восприимчивость. Около 90% пациентов с сахарным диабетом I типа гомозиготны по аллелям DQB1, не кодирующим аспарагиновую кислоту в 57 положении. Раз молекула DQ, и конкретно 57 позиция р-цепи критична для связи антигена и пептида и Т-клеточного ответа, похоже, что различия в присоединении антигена, определяемые конкретной аминокислотой в 57 положении р-цепи DQ, непосредственно содействуют аутоиммунному ответу, уничтожающему инсулин-продуцирующие клетки поджелудочной железы. Тем не менее также важны другие локусы и аллели в МНС, что видно из того, что некоторые пациенты с сахарным диабетом I типа имеют в данной позиции b-цепи DQ аспарагиновую кислоту. Гены, отличающиеся от локусов главного комплекса гистосовместимости класса II при сахарном диабете I типаГаплотип МНС отвечает только за часть генетического вклада в риск сахарного диабета I типа у сибсов пробанда. Семейные исследования показывают, что даже когда сибсы имеют те же гаплотипы МНС класса II, риск болезни составляет приблизительно 17%, что значительно ниже показателя конкордантности у однояйцовых близнецов, равного примерно 40%. Таким образом, в геноме должны быть другие гены, также предрасполагающие к развитию сахарного диабета I типа и различающиеся у однояйцовых близнецов и сибсов, имеющих аналогичные условия окружающей среды. Кроме МНС, предполагают изменения еще в более чем десятке локусов, увеличивающих восприимчивость к сахарному диабету I типа, но надежно подтверждены только три из них. Это вариабельность числа тандемных повторов в промоторе гена инсулина и простой нуклеотидный полиморфизм в гене иммунного регулятора CTLA4 и в гене PTPN22, кодирующем протеин-фосфатазу. Идентификация других генов восприимчивости для сахарного диабета I типа как в пределах, так и за пределами МНС — объект интенсивного исследования. В настоящее время природа факторов негенетического риска при сахарном диабете I типа в основном неизвестна. Генетические факторы сами по себе, тем не менее, не вызывают сахарный диабет I типа, поскольку показатель конкордантности у однояйцовых близнецов составляет не 100%, а только около 40%. До получения более полной картины участия генетических и негенетических факторов в развитии сахарного диабета I типа консультирование по оценке риска остается эмпирическим. – Также рекомендуем “Генетика болезни Альцгеймера. Особенности наследования” Оглавление темы “Генетика заболеваний”:
|
Источник