
Неврологические нарушения при генетических синдромах

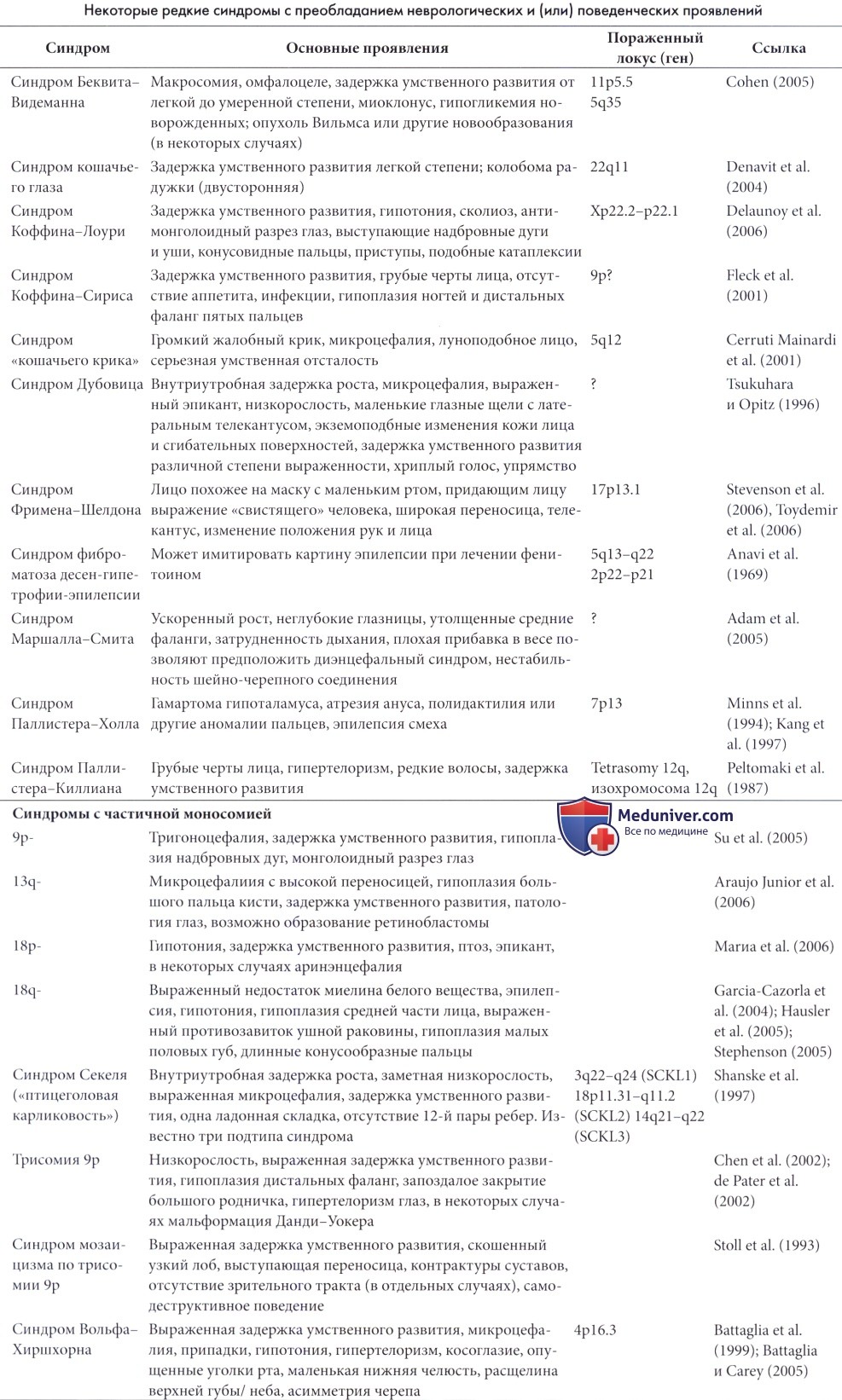
Генетические причины неврологических и умственных отклоненийНеврологические отклонения, в частности нарушения умственного развития и поведенческие отклонения чрезвычайно распространены среди пациентов с хромосомными аномалиями. Частота хромосомных аномалий, обычно приводящих к мальформациям головного мозга, резко повышена среди плодов, погибших в результате выкидыша, и мертворожденных детей. В ходе одного из исследований (Gostason et al., 1991) хромосомные аномалии обнаруживались у 19,2% пациентов с умеренной задержкой умственного развития, по сравнению с 1,9% в контрольной группе. Данные результаты, безусловно, недооцениваются, так как малозаметные хромосомные аномалии, такие как небольшие делеции или транслокации, могут быть выявлены только с помощью современных более сложных методов оценки. Хромосомные аномалии и задержка умственного развития не всегда взаимосвязаны. Аномалии половых хромосом и минимальные аномалии аутосом часто сопровождаются нормальным интеллектом, тем не менее, нельзя сказать, что данные аномалии не приводят к некоторому снижению интеллекта и склонности к развитию поведенческих отклонений. В последние годы стало известно, что классические хромосомные «аберрации» являются только верхушкой айсберга, а неврологические/поведенческие симптомы и признаки являются результатом отсутствия или повреждения особо значимых генов, проявляющегося в форме делеций, дупликаций и других хромосом ных перестроек. Таким образом, традиционное разделение на хромосомные и другие синдромы в настоящее время перестает быть актуальным. В течение последних двух десятилетий стало известно, что ряд заболеваний, обычно генетического происхождения, характеризуется более или менее определенным поведенческим фенотипом. Зарегистрировано несколько сотен синдромов, связанных с хромосомными аномалиями/специфическими генными нарушениями (от одиночных делеций/мультипликаций гена и партеногенетический дисомий до множественного поражения генов определенного участка хромосомы), а их описание можно найти в специализированной литературе (De Grouchy и Turleau, 1984; Schinzel, 1984; Jones, 1988; O’Brien и Yule, 1996). В некоторых случаях, таких как синдром Ангельмана, синдром фрагильной Х-хромосомы, синдром Прадера-Вилли и синдром Вильямса, поведение настолько характерно, что вызывает предположение о наличии у ребенка специфической хромосомной/ генетической аномалии. Зачастую поведенческие симптомы более патогномоничны, чем любые физиологические симптомы или признаки. В настоящее время также известно много дисморфических синдромов с неидентифицированным генетическим дефектом или поражением головного мозга; многие из них проявляются задержкой умственного развития и (или) неврологическими/поведенческими симптомами или признаками. В статьях данного раздела сайта приведены только некоторые из наиболее распространенных и важных с неврологической или поведенческой точки зрения хромосомных/генетических аномалий или дисморфических синдромов. Для удобства они приведены в алфавитном порядке. Другие более редкие синдромы приведены в таблице ниже. Задержка общего и/или умственного развития, степень выраженности которой варьирует от легкой до тяжелой, встречается при многих синдромах с поведенческим фенотипом. Несмотря на то, что общие признаки могут относиться к определенному синдрому, каждому ребенку необходимо индивидуальное обследование и программа лечения в соответствии с его возможностями, учитывая медицинские, социальные и образовательные рекомендации. Более подробные сведения по этой теме приводятся в отдельных статьях на сайте.
– Также рекомендуем “Синдром Ангельмана (синдром счастливой марионетки) – этиология, клиника, диагностика” Редактор: Искандер Милевски. Дата публикации: 4.12.2018 Оглавление темы “Наследственные синдромы в неврологии.”:
|
Источник
Неврологическая симптоматика и гены
Неврологическая симптоматика наблюдается при многих наследственных заболеваниях, и в ряде случаев патологические изменения касаются только нервной системы. Согласно недавним молекулярно-генетическим исследованиям, многие заболевания нервной системы были переклассифицированы в зависимости от лежащего в их основе генетического дефекта.
Заболевания, связанные с делециями генов
Многие неврологические заболевания связаны с полными или частичными делециями генов, например, наследственная полинейропатия с компрессионным параличом.
Заболевания, связанные с дупликацией генов
Некоторые заболевания могут быть связаны с дупликацией генов, примером является наследственная мотосенсорная полинейропатия, при которой у больных развиваются слабость и атрофия дистальных мышц и утрата чувствительности на первых десятилетиях жизни.
Заболевания, связанные с мутацией генов
Мутации генов – наиболее часто встречающиеся генетические дефекты. В основе заболеваний, связанных с мутациями генов, лежит нарушение функции определенного белка или фермента. Примерами таких заболеваний являются семейные формы болезни двигательного нейрона, миодистрофий и миотонических синдромов.
Заболевания, связанные с импринтингом генов
Импринтинг – это дифференциальная экспрессия генов в зависимости от родителя, от которого они были унаследованы. Так, нарушение функции материнских генов на определённых участках хромосом приводит к развитию синдрома Прадера-Уилли (умственная отсталость с ожирением, уменьшением роста), тогда как нарушение функции тех же генов, полученных от отца – к развитию синдрома Ангельмана (выраженная умственная отсталость, атаксия, эпилепсия и аномалия черепа и лица)
Заболевания тринуклеотидных повторов
Патологические тринуклеотидные повторы встречаются при таких заболеваниях как хорея Гентингтона, атаксия Фридрейха.
Наследование психических расстройств и неврологических заболеваний
Недавно в журнале Science была опубликована статья, рассказывающая об аспектах наследования неврологических и психических расстройств. Оказалось, что большинство диагнозов коррелирует с умственными способностями человека, уровнем образования и чертами характера. При этом разные неврологические болезни связаны с наличием разных вариаций в ДНК (то есть практически не наследуются), в то время как многие психические расстройства имеют общие ДНК-варианты.
Что выяснилось:
1. Наследуемость признаков. Когнитивные способности наследуются лучше, чем поведенческие особенности и предрасположенность к болезням. Если сравнивать наследуемость разных заболеваний между собой, то заметно, что чем раньше они проявляются, тем лучше наследуются.
2. Корреляции между заболеваниями. Многие психические расстройства связаны друг с другом, но неравномерно. Корреляция обнаружилась внутри группы шизофрении, биполярного расстройства, тревожного расстройства, большого депрессивного расстройства и синдрома дефицита внимания и гиперактивности.
Вторая такая группа, связанная между собой, включает синдром Туретта, обсессивно-компульсивное расстройство и большое депрессивное расстройство. Шизофрения коррелировала почти со всеми исследованными заболеваниями.
В целом психические и неврологические заболевания оказались практически не связаны друг с другом. Исключение составляет мигрень, у которой обнаружились корреляции с синдромом дефицита внимания и гиперактивности, большим депрессивным расстройством и синдромом Туретта.
По отношению к когнитивным способностям заболевания разделились на две группы:
1. синдромом дефицита внимания и гиперактивности, тревожным и депрессивным расстройствами, а также синдромом Туретта — корреляции когнитивных способностей были отрицательными.
2. нервной анорексией (Anorexia nervosa), расстройством аутистического спектра, биполярным и обсессивно-компульсивным расстройствами — напротив, положительными.
Что же касается дополнительных признаков, то наиболее сильные корреляции выявили между невротичностью и рядом заболеваний (нервная анорексия, мигрень, обсессивно-компульсивное расстройство и др.), а также между «депрессивной триадой» (снижением настроения и ангедонией, нарушением мышления и двигательной заторможенностью) и биполярным расстройством, синдромом дефицита внимания и гиперактивности и т. д.
Судя по полученным данным, с точки зрения генетики две группы заболеваний нервной системы — психические и неврологические — практически между собой не связаны, несмотря на то, что некоторые симптомы их могут быть общими (как, например, психоз при шизофрении и болезни Альцгеймера). Это значит, что их причины и патогенез принципиально различны.
В то же время, психические расстройства сильно коррелируют друг с другом. Это может означать, что существуют какие-то генетические особенности, являющиеся фактором риска сразу для множества заболеваний. Например, склонность к навязчивым идеям может перерастать в бред, сопровождающий многие заболевания, от нервной анорексии и обсессивно-компульсивного расстройства до шизофрении. Но также этот факт может свидетельствовать о том, что наша действующая классификация психических расстройств неточна. Возможно, в основе многих заболеваний, которые сейчас рассматриваются отдельно друг от друга, лежат общие механизмы возникновения. В таком случае, современная классификация отражает лишь общую симптоматику, но не пути развития болезней и требует основательного пересмотра.
Статья опубликована в журнале Science. Источник: DOI: 10.1126/science.aap8757. https://science.sciencemag.org/content/360/6395/eaap8757
Источник
Хромосомные синдромы в неврологии. Синдром трисомии 21-й хромосомы – синдром ДаунаПод хромосомными синдромами подразумевается группа врожденных пороков развития, обусловленных аномалиями числа или структуры хромосом, т. е. избытком или нехваткой тех или иных генов. Хромосомные аномалии могут возникать в гаметах родителей вследствие нерасхождения хромосом в процессе деления, их поломки или перестройки под действием биологически активных физических, химических, а также биологических факторов. В результате зигота уже содержит аномалию, которая наследуется всеми клетками развивающегося из зиготы организма. Если аномалия возникает в процессе дробления зиготы, то в развивающемся организме оказывается два клеточных клона, отличных по кариотипу — хромосомный мозаицизм. Мозаицизм иногда бывает очень сложным вследствие повторных хромосомных нарушений. Фенотипические проявления аномалий хромосом, по-видимому, в большей степени обусловлены больше несбалансированностью кариотипа, чем изменением дозы тех или иных конкретных генов. Поэтому в первую очередь следует иметь в виду семиотику хромосомных синдромов в целом, которая складывается из полного спектра врожденных пороков развития и дизэмбриогенетических стигм. Патогномоничпое их сочетание при определенных хромосомных аномалиях дает картину определенного синдрома. Однако клинический диагноз требует верификации исследованием кариотипа. Хромосомные синдромы подразделяются на синдромы, обусловленные аномалиями аутосом (любой из 22 пар неполовых хромосом) или половых Х- или Y-хромосом. Синдромы, обусловленные аномалиями аутосомСиндромы этой группы характеризуются множественными врожденными пороками развития, глубокой задержкой психического и физического развития, сниженной жизнеспособностью. Их классифицируют в соответствии с порядковым номером хромосом, гены которых присутствуют в избытке или в недостатке.
Синдром трисомии 21-й хромосомы (синдром Дауна)Симптомокомплекс, обусловленный избытком генов, локализованных в 21-й хромосоме, описан в 1866 г. L. Down. Этиологическую значимость хромосомной аномалии при этом синдроме подтвердили J. Lejeune и др. в 1959 г. В 98% случаев синдром обусловлен регулярной трисомией 21-й хромосомы, в 2% —избыточный хромосомный материал транслоцирован на одну из аутосом (чаще из группы D и G). Может быть также мозаицизм. Частота болезни Дауна среди новорожденных, по данным различных исследователей, колеблется в пределах 1 : 290 — 1 : 1935. По мнению J. Oster, среди новорожденных она составляет 1 : 765, а в популяции — 1 : 4000. Частота транслокационных случаев болезни Дауна в популяции достигает 1 : 37 000— 48 000. С увеличением возраста матери возрастает частота случаев рождения детей с болезнью Дауна. Среди женщин старше 45 лет она составляет 1 : 20 — 1 : 45 новорожденных, в то время как среди матерей моложе 20 лет — 1 : 700 новорожденных. Транслокационный синдром чаще встречается у детей, рожденных от молодых матерей. Патологоанатомические изменения характеризуются недоразвитием мозга и внутренних органов: аномальное строение головного (микрогирия, пахигирия) и спинного мозга, атрофия мозжечка, задержка процессов миелинизации, нарушения дифференциации различных отделов центральной нервной системы, гетеротопии, врожденные пороки сердца (дефект межжелудочковой или межпредсердной перегородки) и крупных сосудов, пороки развития кишечника, почек, диафрагмальные и пахово-мошоночные грыжи. Заболевание проявляется с рождения, и диагноз в большинстве случаев ставится уже в родильном доме. Внешний вид новорожденного характерен: череп округлой формы, затылок плоский, косой разрез глаз, эпикант, широкая переносица, пятна Брушфильда на радужной оболочке, яркий румянец на щеках, маленькие прижатые к черепу уши; нос маленький, приплюснутый; недоразвитие верхней челюсти, готическое нёбо, рот полуоткрыт, углы его опущены, язык толстый и покрыт поперечными бороздами, гипертрофия сосочков языка (рис. 55). Пальцы кистей и стоп укорочены, V палец часто искривлен, синдактилия, сандалевидная щель между I и II пальцами на ногах. В 50% случаев выявляются гипоплазия половых органов, эндокринные нарушения. Неврологический статус: выражена общая мышечная гипотония, в результате чего увеличен объем пассивных движений. Новорожденные вялы, адинамичны, крик слабый, болезненпый, нередко нарушены сосание и глотание; безусловные рефлексы угнетены. Аномалия развития голосовых связок является причиной стридорозного дыхания. При наличии порока сердца могут иметь место повторные приступы асфиксии. Благодаря характерным внешним признакам диагноз болезни Дауна поставить не трудно, за исключением случаев, когда основные симптомы нечетко выражены. Характерны дерматоглифические изменения: поперечная ладонная борозда, повышена частота петель на IV и V пальцах, ульнарных петель на I —III пальцах, дуг — на IV пальцах, завитков — на V пальцах, частота этих узоров на других пальцах снижена; высокий трирадиус, частота истинных узоров в III межпальцевом промежутке на левой руке повышена, на правой — снижена. Даже при отсутствии сомнения в диагнозе следует проводить исследования кариотипа с целью выявления транслокационных вариантов, обнаружение которых является показанием для исследования кариотипа родителей. Если один из родителей является носителем сбалансированной транслокации, то последующее деторождение без аптенатального контроля кариотипа плода не показано. При стертых формах болезни Дауна иногда выявляется мозаицизм, когда одна часть соматических клеток содержит 46 хромосом, а другая — 47. Для обнаружения мозаицизма иногда необходимо провести исследование кариотипа не только в лимфоцитах периферической крови, но и в фибробластах кожи. Лечение в период новорожденности сводится к стимулирующей и общеукрепляющей терапии, профилактике интеркурентных заболеваний, рациональному вскармливанию, лечебной гимнастике. В настоящее время с определенным успехом применяют препараты глютаминовой кислоты, церебролизин, тиреоидин, префизон, аминолои, тиамин, пиридоксин, цианокобаламин, кальция пангамат, АТФ, ниамид. Наряду с медикаментозными препаратами большое значение при болезни Дауна придается медико-педагогическим мероприятиям и воспитанию двигательных навыков. – Также рекомендуем “Синдром трисомии 18-й хромосомы. Синдром Эдвардса в неврологии” Оглавление темы “Наследственные синдромы в неврологии”: |

Источник
Геномная классификация в неврологии. Генетическая индексацииИдентификация генов, разработка разнообразных методов ДНК-диагностики и установление феномена генетической гетерогенности наследственных болезней нервной системы позволили на качественно новом уровне решить проблему построения четкой и упорядоченной современной номенклатуры этих заболеваний, приведя к внедрению наиболее совершенного и принципиально нового геномного подхода к классификации. Геномная (генетическая) классификация предполагает определение прямой взаимосвязи между конкретной нозологической формой и повреждением определенного гена, лежащим в основе болезни [Hardy J., HardyGwinn К., 1998]. Ярким примером может служить группа аутосомно-доминантных атаксий, систематизация которых до недавнего времени была чрезвычайно затруднена. В последнее десятилетие было показано, что вариабельность клинико-анатомической картины аутосомно-доминантных атаксий обусловлена генетической гетерогенностью данной группы заболеваний. На сегодняшний день установлено существование, как минимум, 16 самостоятельных генов, локализованных на различных хромосомах, мутации которых обусловливают развитие доминантных атактических синдромов. В результате стала возможной предельно четкая и объективная молекулярно-генетическая классификация доминантных атаксий, основанная на прямой (выявление мутации в конкретном гене) или косвенной ДНК-диагностике (установление сцепления с конкретным хромосомным локусом). По такому же принципу строится в настоящее время классификация наследственных спастических параплегии (картированы на хромосомах 15 локусов данных заболеваний, для 4 из которых идентифицированы мутантные гены), нейрональных цероид-липофусцинозов (6 хромосомных локусов, в том числе 5 идентифицированных генов), семейных форм первичного паркинсонизма (7 локусов, в том числе 3 идентифицированных гена) и других групп наследственных неврологических заболеваний.
В ряде случаев при построении современной классификации генетическая индексация дополняется обозначением типа наследования и некоторых других базовых клинических признаков. Так, в группе аутосомных форм конечностно-поясной прогрессирующей мышечной дистрофии (КПМД) цифровой индекс обозначает тип наследования (1 – аутосомио-доминантный, 2 – аутосомно-рецессивный), а буквенный – конкретный хромосомный локус. Например, символ КПМД 1В обозначает аутосомно-доминантную форму конечностно-поясной мышечной дистрофии, ген которой расположен на хромосоме lql l-q2I, а символ КПМД 2D – аутосомно-рецессивную форму, обусловленную мутацией гена а-саркогликана на хромосоме 17ql2-q21.33. Аналогичным образом, группу наследственных моторно-сепсорных невропатий принято условно подразделять на демиелинизирующие (тип I) и аксональные формы (тип И), в рамках каждой из которых выделяются отдельные генетические подтипы, например: F1MCH 1А (или СМТ 1А -от англ. Charcot-Marie-Tooth) – обусловлена мутациями гена РМР22 на хромосоме 17pll.2; HMCII 1В (СМТ 1В) – мутации гена Р0 па хромосоме 1 q22-23; НМСН 2В (СМТ 2В) – аксональиая форма, сцепленная с хромосомой 3q 13-22 и т.д. Эти примеры ясно показывают, что для успеха ДНК-диагностики и практической реализации геномного подхода к классификации наследственных заболеваний нервной системы чрезвычайно важным является тесное сотрудничество клинициста-невролога и врача лаборатории, осуществляющего мутационный скрининг. Квалифицированный клинико-генеалогический анализ синдрома является краеугольным камнем, на котором основаны постановка показаний к проведению ДНК-диагностики, отбор генов, подлежащих исследованию, последовательность процедур мутационного скрининга, а также адекватная трактовка получаемых результатов. – Также рекомендуем “ДНК-диагностика в неврологии. Локусная гетерогенность” Оглавление темы “Классификация генетических болезней в неврологии”: |
Источник