
Оливопонтоцеребеллярная дегенерация код мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
Названия
Название: Оливопонтоцеребеллярные дегенерации.
Оливопонтоцеребеллярные дегенерации
Описание
Оливопонтоцеребеллярные дегенерации. Наследственные дегенеративные заболевания ЦНС, объединенные сходной локализацией патологического процесса в мозжечке, нижних оливах и мосте головного мозга. Клиника складывается из мозжечкового синдрома, экстрапирамидных расстройств, когнитивных и психических нарушений. Диагностируются оливопонтоцеребеллярные дегенерации на основании анамнеза, генеалогического исследования, данных неврологического и психологического обследования, результатов КТ и МРТ головного мозга. Терапия симптоматическая, включает нейропротекторные и общеукрепляющие фармпрепараты, ЛФК, массаж. Прогноз неблагоприятный.
Дополнительные факты
Прогрессирующий мозжечковый синдром в сочетании с экстрапирамидными нарушениями и психическими расстройствами был описан Дежерином и Томасом в 1900 году. В последующем были выделены несколько форм данной патологии, объединенные в единую группу заболеваний, получившую название «оливопонтоцеребеллярные дегенерации» (ОПЦД). По сути, речь идет о заболеваниях с единой локализацией мультифокальной дегенерации и гибели нейронов. Именно преимущественное расположение дегенеративных процессов легло в основу названия этой группы поражений ЦНС (с латинского oliva — олива, pont(is) — мост, cerebellum — мозжечок). В ряде случаев наблюдается поражение каудальных черепно-мозговых нервов (IX, X, XI, XII пар), реже — передних рогов спинного мозга и проводящих трактов.
Оливопонтоцеребеллярные дегенерации входят в группу дегенеративных поражений ЦНС, к которой относятся болезнь Паркинсона, рассеянный склероз, болезнь Альцгеймера, лейкодистрофии, болезнь Пика, спинальные амиотрофии и мн. Тд Отмечается аутосомное рецессивное и доминантное наследование, спорадические случаи. Возраст манифестации клинических проявлений варьирует в пределах от 11 до 80 лет, наиболее часто дебют происходит в четверной или пятой декаде жизни.
Оливопонтоцеребеллярные дегенерации
Причины
Точные представления о этиопатогенезе ОПЦД пока не сформированы. Поиск генетического субстрата дегенераций привел к выявлению нарушений в локусе 6p22-23 (при дегенерации Менделя) и в локусе 12q23-24 (при дегенерации Фиклера-Винклера) в виде увеличения числа тринуклеотидных повторов. У ряда пациентов наблюдается недостаточность дегидрогеназы глутаминовой кислоты, необходимой для метаболизма глутамата. Последний в качестве медиатора активирует передачу возбуждения от мозжечковой коры к клеткам Пуркинье, аксоны которых формируют эфферентные (нисходящие) мозжечковые тракты. Избыточное накопление глутамата при дефиците дегидрогеназы оказывает нейротоксический эффект, который, возможно, является основной причиной дегенеративных изменений клеток Пуркинье.
Основными морфологическими признаками ОПЦД выступают: асимметричные атрофические изменения белого вещества мозжечковых полушарий и в меньшей степени его червя, дегенерация средней и нижней мозжечковых ножек, глиоз и сморщивание ядер моста и олив. Атрофия коры мозжечка с утратой клеток Пуркинье отмечается на более поздних стадиях ОПЦД. Типична полная интактность верхней ножки, узелка червя (nodulus) и клочка (flocculus) мозжечка. Гистологический анализ пораженных церебральных тканей выявляет дистрофически-дегенеративные изменения нейронов, разрастание глии, демиелинизацию нервных волокон.
Классификация
В настоящее время в клинической неврологии известны 5 основных типов оливопонтоцеребеллярных дегенераций. Отдельно выделяют синдром Шая-Дрейджера, который наряду с оливопонтоцеребеллярной дегенерацией включает диффузную церебральную атрофию и дегенерацию стрионигральных структур.
Tип I Менделя. Аутосомно – доминантная ОПЦД с дебютом после 11 лет и до 60 – летнего возраста. Характерна мозжечковая атаксия, гиперкинезы, гипотония мышц, дисфагия. Реже встречаются пирамидные расстройства (парезы конечностей, гипестезия).
Tип II Фиклера. Винклера – аутосомно – рецессивная ОПЦД, манифестирующая с третьей декады жизни до 80 – летнего возраста. Протекает без нарушений чувствительности и парезов. Глубокие рефлексы сохранены.
Tип III ОПЦД с ретинальной дегенерацией. Аутосомно – доминантная форма, поражающая преимущественно лиц молодого возраста. Характеризуется мозжечковым синдромом и гиперкинезами в сочетании с прогрессирующим падением зрения вследствие пигментной ретинопатии.
Tип IV Шута. Хайкмана – аутосомно – доминантная ОПЦД детского и молодого возраста. Типичные для всех ОПЦД мозжечковые расстройства сочетаются с поражением каудальных черепных нервов (бульбарные симптомы) и задних столбов спинного мозга (нарушение глубокой чувствительности).
Tип V ОПЦД с деменцией, экстрапирамидными знаками и офтальмоплегией. Наследуется аутосомно-доминантно. Представляет собой комбинацию указанных в названии синдромов и мозжечковой атаксии.
Симптомы
Базисным клиническим проявлением ОПЦД является мозжечковая атаксия. Дебют заболевания знаменуется появлением легкой неустойчивости, неуклюжести при беге и быстрой ходьбе. Прогрессирование этих симптомов приводит к выраженным расстройствам походки и статики. Ходьба затруднена, сопровождается падениями, во избежание которых пациенты широко расставляют ноги во время ходьбы. Грубые нарушения равновесия обуславливают колебания туловища пациента, когда он стоит или сидит. Позднее присоединяется дискоординация в конечностях: адиадохокинез, гипер- и дисметрия, крупноразмашистый почерк. Атаксия в конечностях сопровождается дрожанием головы и интенционным тремором. Наблюдается горизонтальный нистагм. Одновременно с атаксией в конечностях появляется типичная мозжечковая дизартрия, т. Н. «скандированная речь». Как правило, происходит повышение рефлексов, в отдельных случаях — их понижение.
Возможны расстройства глотания (дисфагия), гиперкинезы, симптомы вторичного паркинсонизма, лицевой парез, пирамидная недостаточность. При поражении ядер каудальных черепных нервов возникает офтальмоплегия, бульбарный паралич. В большинстве случаев оливопонтоцеребеллярные дегенерации протекают с недержанием мочи. В эмоциональной сфере преобладает снижение: вялость, безынициативность, тупость. Обычно отмечаются значительные когнитивные нарушения и деменция. Характерны психические расстройства: галлюцинаторный синдром, депрессия, фобические расстройства, эпизоды психомоторного возбуждения, спутанность сознания. Иногда их появление предшествует развитию мозжечковой атаксии.
Вялость. Галлюцинации. Тремор. Тремор головы.
Диагностика
Постановка диагноза требует сопоставления времени и симптомов дебюта заболевания, данных неврологического статуса (сочетание мозжечковых нарушений с гиперкинезами) и нейропсихологического обследования (наличие когнитивного снижения, отклонения в эмоциональной сфере) с результатами нейровизуализации.
Дифференциальная диагностика
Дифференцировать оливопонтоцеребеллярные дегенерации необходимо от атаксии Пьера-Мари и атаксии Фридрейха, опухолей мозжечка, прогрессирующих вариантов рассеянного склероза, дисметаболических заболеваний с мозжечковым синдромом (например, болезни Рефсума).
Компьютерная томография малоинформативна, поскольку определяет преимущественно неспецифические изменения церебральных структур: расширение желудочков и субарахноидальных пространств. Специфичным признаком, регистрируемым при помощи КТ головного мозга, является уменьшение толщины передней мозжечковой ножки. Более полно диагностировать оливопонтоцеребеллярные дегенерации позволяет МРТ головного мозга. С ее помощью можно визуализировать атрофические изменения в мосте и продолговатом мозге. Для определения типа наследования патологии необходима консультация генетика и генеалогическое исследование. При подозрении на ОПЦД I или II типов возможна ДНК-диагностика. Пациенты с нарушением зрения нуждаются в консультации офтальмолога.
Лечение
Специфическая терапия ОПЦД пока не найдена, поэтому неврологами осуществляется симптоматическое лечение, т. Е. Направленное на уменьшение конкретных клинических проявлений. Показаны нейрометаболиты и общеукрепляющие средства: витамины гр. В, витамин С и тд Для нормализации мышечного тонуса проводится массаж и ЛФК. С целью уменьшения выраженности нарушений координации рекомендованы специальные упражнения для ее тренировки. При синдроме паркинсонизма показаны центральные холинолитики: диэтазина гидрохлорид, тригексифенидил.
Прогноз
Несмотря на проводимое лечение, оливопонтоцеребеллярные дегенерации имеют неуклонно прогрессирующее течение. Длительность заболевания в среднем колеблется в пределах 10-15 лет, иногда достигает 20 лет. Причиной летального исхода, как правило, являются интеркуррентные инфекции (застойная пневмония, сепсис).
Источник
Оливопонтоцеребеллярные дегенерации (ОПТСД), как отельный вид заболевания был описан более 100 лет назад. В течение всего периода болезнь изучалась, разрабатывались методы диагностики и лечения.
Сегодня известно, что оливопонтоцеребеллярная дегенерация передается по наследству. Болезнь поражает центральную нервную систему. Локализация атрофии – мозжечок, мост и нижние оливы (отделы головного мозга).
Основным проявлением служит изменение в привычном поведении – отмечаются атаксия, экстрапирамидные нарушения. Этот вид заболевания характеризуется длительностью протекания, но фиксируется оно в зрелом возрасте. В процессе изменений, происходящих в мозге человека, отмечается гибель нейронов, соответственно, нарушаются частота проведения сигналов от них в ЦНС.
Также дегенеративно-дистрофические процессы, которые происходят на всем протяжении развития заболевания, поражают кору мозжечка, мост и оливы мозга.
Именно поэтому наблюдаются характерные изменения в психике и возможностях человека, включая физические. В редких случаях во время  диагностики отмечается поражение таких участков, как передние рога спинного мозга и его проводящие пути. Также болезнь может затрагивать каудальные черепно-мозговые нервы (9-12 пар). К нарушениям, которые связанны с оливопонтоцеребеллярной дегенерацией, относят такие заболевания:
диагностики отмечается поражение таких участков, как передние рога спинного мозга и его проводящие пути. Также болезнь может затрагивать каудальные черепно-мозговые нервы (9-12 пар). К нарушениям, которые связанны с оливопонтоцеребеллярной дегенерацией, относят такие заболевания:
- болезнь Альцгеймера;
- рассеянный склероз;
- болезнь Пика.
Наследование заболевания происходит по рецессивному и доминантному пути, а также известны случаи спорадического наследования.
Состояние больного может выражаться в потери ориентации, провалах в памяти, полном или частичном склерозе, нарушениях в походке, скованностью движений, снижении двигательной активности в целом и тонусе организма, апатии, потери способности концентрировать внимание.

Статистические данные
По имеющимся сведениям большая часть случаев заболевания отмечается у людей пожилого возраста – после 60-65 лет – 75% случаев.
Однако дегенерация может начать развиваться и в более раннем возрасте – в период 30-40 лет, а в случае наследования и у детей в возрасте 10-11 лет (2-3%).
Именно поэтому проходить обследование необходимо не только по достижению пожилого возраста, но и в 20-40 лет, а в случае врожденных нарушений – постоянно на протяжении всей жизни.
Причины развития болезни
Важной особенностью болезни является тот факт, что точные причины ее возникновения и развития, если не отмечено случаев заболевания в семье, не известны медицине даже спустя век после первого описания. Отмечаются нарушения в локусах генов, выражающихся в увеличении числа тринуклеотидных повторов.
Среди остальных возможных причин, которые могут привести к появлению характерных нарушений отмечаются асимметричные атрофические изменение в белом веществе. Атрофия коры мозжечка отмечается на поздних стадиях заболевания. Для того чтобы узнать точные причины, необходимо пройти полное исследование.
Разновидности течения
Выделяют несколько основных видов оливопонтоцеребеллярной дегенерации:
- тип Менделя;
- тип Фиклера-Винклера;
- атрофия с ретинальной дегенерацией;
- тип Шута-Хайкмана;
- атрофия с деменцией.
Также в отдельную категорию можно включить синдром Шая — Дрейджера, который характеризуется следующими основными по частоте проявления симптомами:
- вегетативные расстройства;
- мозжечковые нарушения и атаксия (разной степени выраженности);
- поражение в области базальных ганглиев.
Тип Менделя имеет аутосомно-доминантный механизм наследования и следующие проявления:
- медленное и неяркое развитие заболевания, что провоцирует его развитие и переход в запущенную стадию;
- возраст проявления первых симптомов — с детства и до 60 лет;
- атаксия;
- снижение мышечного тонуса (иногда значительное, человек не может выполнять обычных дел);
- нарушение речи (проявляется от слабого до сильного);
- дрожание и подергивание в руках (тремор);
- нарушение в процессе глотания;
- гиперкинезы;
- нарушения, связанные с движением глаз (редко).
Оливопонтоцеребеллярная дегенерация Фиклера-Винклера проявляется следующими признаками:
- наследуется аутосомно-рецессивным способом;
- возраст появления симптомов и развития заболевания на протяжении всей жизни, начиная с 20 лет;
- атаксия конечностей.
Если диагностируется этот вид, то нарушения двигательной активности не прослеживается.
Тип с ретинальной дегенерацией имеет следующие симптомы:
- аутосомно-доминантный принцип передачи новому поколению;
- страдают молодые люди и дети;
- атаксия;
- экстрапирамидные нарушения;
- изменения в остроте зрения (значительное падение), появляется нарушение по причине пигментации сетчатки.
Вид Шута-Хайкмана отмечается следующими симптомами и проявлениями:
- аутосомно-доминантный — основной метод наследования;
- проявления и развитие в молодом (20-30 лет) или детском возрасте;
- атаксия;
- характерный для этого вида признак — лицевой паралич;
- нарушения глотания (иногда сильные);
- дефекты речи;
- вибрационные нарушения.
Тип с деменцией характеризуется следующими признаками:
- аутосомно-доминантное наследование;
- проявление и развитие — в среднем возрасте, но не позднее 40 лет;
- нарушение интеллекта (иногда очень сильное);
- экстрапирамидные расстройства;
- атаксия.
В целом симптоматика схожа, но имеющиеся отличия дают возможность врачам ставить правильный диагноз и назначать наиболее действенное лечение.
Общие симптомы
Общий симптом для всех видов оливопонтоцеребеллярных дегенераций – это атаксия. В начале у человека отмечается легкая неустойчивость, затем при быстрой ходьбе пациент испытывает дискомфорт, выражающийся в неуклюжих движениях. Дополнительные визуальные нарушения походки:
- частые падения;
- широко расставленные во время ходьбы ноги;
- колебания тела из стороны в сторону (нарушение работы мозжечка);
- неустойчивое положение во время сидения на стуле.
Поздние симптомы (болезнь прогрессирует):
- крупный почерк;
- дисметрия;
- дрожание в конечностях (тремор);
- дрожание головы;
- понижение или повышение рефлексов;
- нарушение речи.
Также к общей симптоматике можно отнести:
- нарушение глотания (отмечается в большинстве случаев);
- пирамидная недостаточность.
 Лицевой парез не является общим для всех видов симптомом, в отличии от недержания мочи.
Лицевой парез не является общим для всех видов симптомом, в отличии от недержания мочи.
Человек почти всегда становится вялым и апатичным, теряет интерес к происходящим вокруг него событиям. Нередко фиксируется заторможенность и даже тупость. Также болезнь сопровождается слабоумием в разной степени выраженности, появляются депрессивные состояния, страхи и фобии, которые ранее отсутствовали. Галлюцинации и спутанность сознания – характерные симптомы этого заболевания.
Если болезнь находится в запущенном состоянии , то человек постепенно теряет возможность сначала ходить, а затем и перестает выполнять простейшие действия по уходу за собой. Нередко в этот период у пациентов ослабевает иммунитет, что приводит к развитию сопутствующих заболеваний.
Диагностические критерии
Для того чтобы диагностировать заболевание, а затем точно узнать его тип, потребуется пройти несколько обследований. Это необходимо для получения объективной и достоверной информации. Одним из обследований является неврологический статус. Также осуществляется исследование нейропсихологических данных.
Все полученные результаты дают врачу возможность выяснить, имеются ли у пациента мозжечковая дегенерация, присутствует ли рассеянный склероз и другие симптомы оливопонтоцеребеллярной дегенерации.
Объективную информацию можно получить, пройдя обследование с помощью компьютерной томографии мозга, а затем и МРТ мозга. Для определения типа заболевания потребуется консультация со специалистом в области генетики.
Дополнительно может потребоваться диагностика ДНК. Если отмечается значительное ухудшение зрения, то к диагностике прибавляется консультация офтальмолога.
Комплекс мер — что можно сделать?
Специального действенного лечения от этого заболевания не создано. Сегодня проводятся мероприятия для поддержания показателей в усредненных и приближенных к норме значениях. В большинстве случаев проводится симптоматическое лечение с уклоном в неврологию.
Также делается упор на симптоматику, присутствующую в момент диагностики.
Основные лекарственные препараты – нейрометаболиты, холинолитики, а также общеукрепляющие средства. Для поддержания оптимального мышечного тонуса проводится лечебный массаж и занятия ЛФК. Также используются такие медикаментозные препараты:
- глутаминовая кислота;
- Церебролизин (уколы);
- Аминалон;
- Прозерин.
Активные занятия на свежем воздухе и простые прогулки входят в комплекс мероприятий по лечению болезни. Если отмечаются нарушения зрения, то в терапию включают соответствующие препараты.
Значительного улучшения в состоянии здоровья человека отмечаться не будет. Однако проводимая терапия позволит поддерживать показатели в пределах допустимых продолжительное время, так длительность заболевания в среднем составляет 12 лет, известны случаи, когда пациенты могли бороться с подобными заболеванием на протяжении 20 лет. Основная причина смерти – пневмония или сепсис, как сопутствующие заболевания.
Источник
Одной из дегенеративных болезней является мультисистемная атрофия головного мозга. Она трудно диагностируется. В ходе обследования надо применять МРТ, ставить пациента на учет невролога с регулярным наблюдением врача. В лечении применяют методы физиотерапии, симптоматические лекарства. С началом прогрессии болезни вскоре нужен будет пожизненный уход за больным.
О заболевании
Множественной системной (мультифокальной, мультисистемной) атрофией называют обширную дегенерацию глиальных клеток во всех структурах головного мозга (ГМ). Сокращенно болезнь записывают МСА или MSA. Мультисистемная атрофия быстро прогрессирующее тяжелое заболевание, при котором в патологический процесс часто вовлекается спинной мозг, приводя к уменьшению тканей центральной нервной системы инвалидности.
Дегенерация провоцирует появление:
- слабоумия;
- дрожательного паралича — синдром паркинсонизма;
- дисфункциивегетативной нервной системы;
- атаксии мозжечка — расстройство двигательной и речевой функции;
- недостаточности пирамид — нарушается работа ЦНС.
Справка! МСА является редкой неизлечимой патологией, встречается преимущественно у мужчин. Заболеваемость составляет менее 6 случаев на 100000 населения.
Во время развития болезни поражаются подкорковые ядра (базальные ганглии) внутри полушарий ГМ.Они расположены между промежуточным мозгом и лобными долями. Состоят из полосатого тела, серого и белого вещества, нигральной (черной) субстанции, субталамического ядра. На гистологическом исследовании в олигодендроглиоцитах обнаруживаются накопления альфа-синуклеина во включениях цитоплазмы. Это несвойственно составу клеток нейроглии.
В результате дегенерации подкорковых ядер нарушается:
- координация движений;
- регуляция мышечного тонуса;
- чувствительность восприятия зрительных, слуховых и прочих раздражений;
- регулирование трофики, обмена, дыхания, мочеиспускания, иных вегетативных функций;
- выработка рефлексов, память, другая регуляция высшей нервной деятельности.
МСА чаще выявляют у людей старше 50 лет, которые работали с вредными и токсическими веществами, пестицидами, формальдегидом, растворителями. В группе риска состоят пациенты с болезнью Паркинсона, Альцгеймера. Опасность заключается в том, что патология вызывает необратимый процесс в мозге с гибелью нервных клеток. С момента появления симптомов длительность жизни не превышает 15 лет. Смерть чаще наступает вследствие нарушения дыхания или сепсиса.
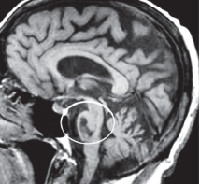
 Рисунок 1. Отображение мозга при томографии
Рисунок 1. Отображение мозга при томографии
До 2016 года в МКБ―10 мультисистемная атрофия была причислена к коду G90.3 под названием «полисистемная дегенерация». Сейчас в справочнике остался паркинсонический тип МСА под шифром G23.2 «MSA-P» и мозжечковый – G23.3 «MSA-C». В МКБ-10-КМ патология отмечена как «мультисистемная дегенерация ВНС (вегетативной нервной системы)» под номером G90.3.
Причины развития
Ученые продолжают изучать причины, механизм развития и провоцирующие факторы МСА. Однозначного подтверждения нет о наследственной предрасположенности к развитию болезни. У ребенка врожденная атрофия ГМ бывает при злоупотреблении женщины во время беременности лекарствами, спиртным, наркотиками.
В ходе диагностики врачи выявляют возможные причины мультисистемнойатрофии:
- болезнь Паркинсона либо Альцгеймера;
- контакт с нейротоксическими веществами;
- изменчивость гена «α-синуклеин»;
- отравление алкоголем, наркотиками у людей с зависимостью;
- травма головного и/или спинного мозга;
- гипоксия тканей ГМ.
Патогенез плохо поддается изучению, поскольку неизвестны точные причины мультисистемной атрофии. В ходе исследований обнаружено накопление тау-протеинав пораженных олигодендроглиоцитах. Это выявляют в мозжечке, пирамидах, коре ГМ, рогах спинного мозга в области грудного и крестцового отдела хребта. Одновременно повреждаются дофаминовые рецепторы, черная субстанция, скапливаетсяα-синуклеин в нейроглиальных клетках.
Справка! МСА характерно асимметрическое уменьшение белого вещества, нарушение передачи нервных импульсов. Нейроны страдают меньше олигодендроглиоцитов.
Классификация
Медики выделяют три формы МСА в зависимости от того, какой синдром выявляется ведущим. Если это невозможно установить, пациенту диагностируют смешанный тип болезни.
Классификация патологии:
| Ведущий симптом | Тип мультисистемной атрофии | Отличительные черты |
| Паркинсонизм | Стриатонигральный тип МСА | Замедленные движения, лицо маскообразное, застывание в одной позе, симптом «воздушная подушка», тремор конечностей, согнутость суставов, снижение подвижности. Дегенерации больше подверженстриатум, черная субстанция. |
| Вегетативная недостаточность | Синдром Шая-Дрейджера | Дисфункция желез и органов, тазовые нарушения, гипотензия, храп, апноэ. |
| Мозжечковая атаксия | Оливопонтоцеребеллярный тип МСА | Ухудшение равновесия, нарушение мелкой моторики, непроизвольное движение глазных яблок, мышечная слабость. Дегенерации больше подвержен мозжечок, мост, оливы. |
В классификаторах врачи предлагают убрать синдром Шая-Дрейджера, поскольку вегетативная недостаточность сопровождает все формы МСА. В МКБ-10 указан только мозжечковый и паркинсонический тип болезни.
Симптомы
Первый признак МСА – начало прогрессирования болезни в старшем возрасте после 45 лет. Симптомы развивается быстро. У большинства людей сразу проявляется паркинсонизм, двигательные нарушения. В 40% случаев дегенерация стартует с вегетативной дисфункции.
Изменения в начальной стадии пациентом не всегда замечается. Среди первых признаков указаны тазовые нарушения: эректильная дисфункция, трудности с мочеиспусканием или дефекацией, недержание мочи/кала. Каждый пятый заболевший с момента прогрессирования мультисистемной атрофии начинает падать. Причиной считается ортостатическая гипотензия, мышечная слабость, дисфункция мозжечка.
Справка! Прогрессированию МСА характерно присоединение к ведущему синдрому других симптомокомплексов. То есть, у человека одновременно проявляется вегетативная недостаточность в сочетании с признакамипаркинсонизма и мозжечковой атаксии.
Отличия симптомов разных типов МСА
Первичные симптомы зависят от класса мультисистемной атрофии. При стриатонигральном типе МСА сразу заметны признаки болезни Паркинсона. Вначале организм откликается на лечение леводопой, затем эффективность лекарств теряется, усугубляются вегетативные расстройства.
Первичные признаки паркинсонического типа мультисистемной атрофии:
| Симптом | Пояснение |
| Брадикинезия | Все произвольные движения замедляются. Человек медленнее ходит, говорит, пишет, читает вслух. Координация движений и речи сохраняется. Длительный разговор или необходимость движения вызывает быструю утомляемость. |
| Ригидность | Скованность движения, напряжение мышц, отвечающих за сокращение, разгибание. Подбородок почти касается ключичной зоны. В горизонтальном положении на спине голова не лежит на подушке, но это исчезает после засыпания. Конечности полусогнуты в крупных суставах, туловище сгибается вперед, позвоночник сутулый. При пассивном движении конечностью (выполняет доктор) под пальцами врач ощущает вязкое сопротивление мышц. |
| Постуральная неустойчивость | Человек не может сохранить равновесие. Это не связано с ортостатической гипотонией, потемнением в глазах, гипертензией. |
| Тремор | Мышцы туловища, шеи, рук, ног дрожат во время движения или покоя. Тремор исчезает, когда пациент выполняет противоположное действие. То есть, перестает либо начинает двигаться. |
При оливопонтоцеребеллярном типе МСА на первом плане стоят симптомы мозжечковой дисфункции. Пациент начинает семенить (уменьшается длина шага). Отмечается шаткость походки, скованность мышц, ухудшение общей координации движений и мелкой моторики. Тремор усиливается при приближении к цели движения. Затрудняется смена быстро чередующихся действий.
При мозжечковом типе мультисистемной атрофии проявляется дизартрия и глазодвигательная (окуломоторная) дисфункция. Их симптомы:
- Приглушенность голоса;
- Растянутое произношение слов;
- Скандированная речь;
- Нарушение модуляция звука, фонации, дыхания во время произношения;
- Ритмичное непроизвольное движение глазных яблок (нистагм).
Синдром Шая-Дрейджера при МСА проявляется расстройством функций тазовых органов, желез. Бывает обморок, коллапс из-за падения давления. К признакам относят нарушение мочеиспускания, опорожнения кишечника, снижение слюнотечения, слезотечения, потоотделения. Отмечается во время сна движения глаз, разговор, кратковременная остановка дыхания. У мужчин ухудшается эрекция, развивается импотенция.
Справка! Прогрессирование МСА проявляется усугублением симптомов 1―3 типов мультисистемной атрофии. Клиника дополняется слабоумием, параличом или парезом, неадекватным поведением, осложнениями дегенерации.
Методы диагностики
Обследоваться надо у невролога. Для постановки диагноза нужно динамическое наблюдение пациента с применением церебрального МРТ. В случае противопоказаний проводят компьютерную томографию ПЭТ, ОФЭКТ.
В начале развития МСА МРТ не покажет атрофических изменений мозговой ткани, но поможет исключить опухоль, энцефалит, рассеянный склероз. Через 1―3 года интенсивной прогрессиивыявляют расширение IV желудочка, выраженную дегенерацию подкорковых ганглий, нижней половины моста, мозжечка, скорлупы.
На осмотре невролог оценивает наличие вегетативной недостаточности в комбинации спаркинсонизмоми/или дисфункцией мозжечка.
Мультисистемная атрофия не подтверждается, если:
- МСА начала развиваться до 30 либо после 75 лет;
- вегетативная недостаточность не сочетается ни с мозжечковой дисфункцией, ни с паркинсонизмом;
- патология также есть у близких родственников (семейный анамнез);
- у пациента выявлена деменция, признаки похожей на МСА болезни;
- лечение паркинсонизма эффективно лекарствами леводопы.
Для постановки диагноза ортостатической пробой исследуют функции вегетативной нервной системы. Нарушение работы тазовых структур выявляют при проведении электромиографии сфинктеров.
На развитие мультисистемной атрофии указывает наличие:
- ортостатической гипотензии — снижение давления после принятия вертикального положения;
- нерегулярного тремора;
- холодности стоп и кистей, скованности их сочленений;
- храпа, который вновь появился либо усилился;
- плача или смеха, несоответствующих переживаемой эмоции;
- тяжелых нарушений речи, голоса — дизартрия, дисфония;
- затрудненных вдохов — инспираторная одышка;
- недержания мочи;
- нарушений эректильной функции;
- кривошеи с наклоном головы к груди — антероколлис;
- непроизвольных движений мимических мышц, языка — орофациальная дистония;
- искривления позвоночника в грудопоясничном переходе с наклонением туловища вперед — камптокормия;
- учащения случаев падения;
- мозжечкового синдрома/паркинсонизма + вегетативной недостаточности.
Справка! МСА достоверно подтверждается патоморфологическим исследованием нейроглии. При жизни биоматериал с глиальными клетками получают посредством биопсии мозга или ткань изымает патологоанатом во время вскрытия.
Лечение

Людей с МСА лечат препаратами симптоматической терапии. Они помогают убрать выраженность паркинсонизма, мозжечковой атаксии и других признаков дегенерации головного мозга.
В симптоматическое лечение мультисистемной атрофии включают:
- медпрепараты леводопы;
- вазоактивные средства;
- нейрометаболические лекарства;
- массаж, водные процедуры, ЛФК, другие методы физиотерапии;
- диету с соблюдением нормы потребления соли;
- немедикаментозные способы устранения ортостатистической гипотензии.
Паркинсонизм в начальных этапах прогрессии МСА лечат комбинированными средствами леводопы с бенсеразидом, карбидопой. Его заменяют при плохой переносимости или неэффективности препаратами альтернативной терапии. При атрофии всех типов используют агонисты дофаминовых рецепторов, лекарства с веществом амантадин.
Проявления мозжечковой атаксии убирают методами физиотерапии. Для облегчения состояния МСА назначают средства с клоназепамом, габапентином, буспироном, пропранололом и прочими веществами.
При тазовых нарушениях на фоне атрофии используют лекарства:
- Силденафил (при эректильной дисфункции);
- Макрогол, слабительные средства (в случае запоров);
- Антагонист альфа1-адренорецепторов + холинергический препарат (задержка мочи);
- Антихолинергическое средство (непроизвольное или болезненное мочеиспускание).
При нарушениях функций мочевого пузыря дополнительно к лекарствам показана периодическая либо постоянная катетеризация органа. Из альтернативных препаратов применяют уколы ботулотоксина. Эти инъекции также назначают для лечения дыхательных расстройств, каптокормии, слюнотечения, дистонии на фоне МСА.
При ортостатической гипотензии показано спать на кроватях с возвышением изголовья, пить много воды, богатой минералами. Надо носить компрессионные чулки. Нельзя переедать, резко вставать после пробуждения. Рекомендуется обучиться изометрическим маневрам. Прописывают минералокортикоиды, гипертензивные средства с мидодрином.
При нарушениях дыхания показана вентиляция легких либо трахеостомия. Камптокормию лечат методами физиотерапии. Лекарствами с зопиклономили клоназепамом устраняют расстройства сна из-за МСА. При депрессии прописывают селективный ингибитор обратного захвата серотонина (группа антидепрессантов).
Справка! Клиницисты продолжают искать новые методы лечения мультифокальной атрофии. Этиологическую терапию нет возможности применять, пока невыяснены причины и механизм развития МСА.
Осложнения
К последствиям прогрессирования МСА относят урогенитальные инфекции, цистит, уретрит, воспаление почек. Бактерии могут проникнуть в кровь и вызвать сепсис. На фоне дыхательных нарушений чаще развивается пневмония, ночное апноэ.
При мультисистемной атрофии возможно поражение продолговатого мозга. Это осложняется расстройством глотательной функции, вызывает смерть из-за паралича дыхательного центра или асфиксии. Вследствие дегенеративных процессов нарушается также работа сердца, сосудистой системы, мозговое кровообращение.
Прогноз и продолжительность жизни
МСА – неизле?