
Презентация синдром марфана скачать бесплатно

1. Синдром Марфана
Презентацию подготовила:
Храмова Нелли
МБФ, 5 курс, группа 15-01
2.
Синдром (болезнь) Марфана — аутосомно-доминантное
заболевание из группы наследственных патологий
соединительной ткани. Синдром вызван мутацией гена,
кодирующего синтез гликопротеина фибриллина-1, и
является плейотропным. Заболевание характеризуется
различной пенетрантностью и экспрессивностью. В
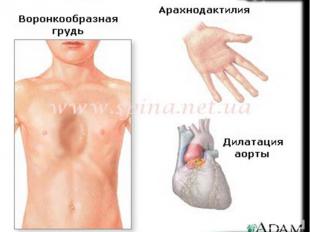
классических случаях лица с синдромом Марфана высоки
(долихостеномелия), имеют удлинённые конечности,
вытянутые пальцы (арахнодактилия) и недоразвитие жировой
клетчатки. Помимо характерных изменений в органах опорнодвигательного аппарата (удлинённые трубчатые кости скелета,
гипермобильность суставов), наблюдается патология в органах
зрения и сердечно-сосудистой системы, что в классических
вариантах составляет триаду Марфана.
3.
История
• Впервые признаки заболевания были описаны в 1875 году
американским офтальмологом Э. Вильямсом, описавшим эктопию
хрусталика у брата и сестры, которые были исключительно высокими
и имели гипермобильные суставы от рождения. В последующие годы
эта болезнь наблюдалась французским профессором
педиатрии Антуаном Марфаном, который представил в 1896 году
клиническое наблюдение 5-летней девочки Габриэль с необычными,
непрерывно прогрессирующими аномалиями скелета, и дал
патологии своё имя.
• Позднее выяснилось, что в действительности девочка страдала
врождённой контрактурной арахнодактилией.
• Американский генетик Виктор Маккьюсик открыл этим синдромом
новую нозологическую страницу наследственных заболеваний
соединительной ткани.
4.
• Синдром Марфана — редкое заболевание с
классическим менделевским наследованием.
Распространённость в популяции составляет порядка 1 на
5000. Синдром диагностируется во всем мире, в любых
этнических группах.
• Существует интересный факт, что первая девушка модель
Лесли Хорнби, которая послужила прототипом образа
всех моделей, имела синдром Марфана. Как, установлено,
что ряд всемирно известных людей страдали синдромом
Марфана, среди них следует упомянуть президента США
А. Линкольна и великого скрипача Паганини.
5. kk
6.
7.
Причины и генетика патологии
Непосредственная причина патологии в 95% случаев – это мутация в гене,
которые кодирует строение фибриллина-1 и/или фибриллина-2.
Локализируется мутация гена FBN1 и FBN2 в хромосоме 15 и 3.
Патологические изменения в одном и том же локусе могут обуславливать
разнообразные клинические проявления от стертой формы с поражением
одной из систем организма до классической развернутой.
Фибриллин – это основа эластических волокон соединительной ткани
гликопротеиновой природы. Он составляет каркас межклеточного
вещества, сосудистых стенок, хрящей, хрусталика глаза и многих других
органов и тканей. В случае наличия описанной мутации у пациента
соединительная ткань отличается повышенной способностью к
растяжению, становится менее прочной и выносливой к механическим
воздействиям, что и становится причиной клинических проявлений
синдрома.
Примерно в 5% случаев непосредственной причиной синдрома Марфана
(атипичные формы патологии) является точковая мутация гена, который
кодирует строение α2-цепи коллагена первого типа.
При вступлении в брак 1 больного и 1 здорового родителя вероятность
рождения больных детей 50%.
8.
9. Классификация
В зависимости от выраженности симптомов:
• стертая форма – признаки патологии мало выражены и могут оставаться
незамеченными на протяжении всей жизни, как правило, изменения
касаются не более 2 систем органов;
• клинически выраженная форма – симптомы патологии хорошо заметны и
встречаются более чем в 2 системах органов.
В зависимости от генетического фактора:
• семейная форма диагностируется в случаях, когда болезнь передается по
наследству;
• спорадическая форма определяется тогда, когда патология обусловлена
новой спонтанной мутацией у индивида и при этом не встречается у его
родственников.
Характер течения:
• прогрессирующий;
• стабильный.
10.
11. Опорно-двигательный аппарат
Патологическая соединительная ткань обусловливает развитие ряда специфических
фенотипических признаков и деформаций скелета у пациентов с данной патологией.
Как правило, внешне пациенты с синдромом Марфана выглядят достаточно специфически.
Для них характерно:
астеническое телосложение;
высокий рост;
плохое развитие подкожной жировой клетчатки, из-за чего люди выглядят худощавыми;
очень длинные верхние и нижние конечности при относительно коротком туловище;
череп вытянутый (долихоцефалический);
удлиненные пальцы – паукообразные (арахнодактилия);
лицо узкое, вытянутое по вертикали;
готическое верхнее небо;
недоразвитие скул;
выступающая нижняя челюсть (прогнатизм);
неправильный рост (скученность) зубов и патологический прикус;
гипермобильность суставов, их «разболтанность;
глубоко посажены в черепе глаза.
По мере роста ребенка могут появляться различные деформации скелета. Чаще всего появляются
искривления позвоночного столба. У пациентов диагностируют сколиоз, патологический
кифоз и лордоз. Для пациентов также характерна остеопения (снижение минеральной
плотности костей) и частые патологические переломы костей на ее фоне, а также склонность
к привычным вывихам, например, плеча.
12.
13.
14.
15. Сердечно-сосудистая система
Среди поражений кардиоваскулярной системы при СМ чаще всего
встречаются:
пролапс створок митрального клапана с регургитацией или без
миксаматоз сердца;
дилятационная кардиомиопатия с развитием сердечной недостаточности;
аневризмы аорты и других сосудов (мозговых, почечных, пр.);
расширение легочной артерии и различных отделов аорты.
Именно кардиоваскулярные патологические изменения при СМ определяют
прогноз и продолжительность жизни пациентов. Примерно 90% всех
пациентов с данной генетической патологией умирают в возрасте 40-50 лет
вследствие таких осложнений, как расслоение и разрыв аневризмы аорты,
других сосудов, прогрессирующей недостаточности сердца вследствие
дилатации его камер и изменений клапанного аппарата.
При наличии СМ возможно наличие врожденных пороков сердца у детей.
Чаще всего встречаются коарктация аорты, стеноз (сужение) легочной
артерии, дефект межжелудочковой и межпредсердной перегородки.
Также такие пациенты склонны к различным сердечным аритмиям,
среди которых и опасные для жизни (мерцательная аритмия,
желудочковая тахикардия и экстрасистолия), к инфекционному
эндокардиту.
16.
Аневризмы аорты и их разрыв чаще всего становятся причиной смерти пациента с
синдромом Марфана
17. Орган зрения
Патологические изменения глаз являются весьма
характерными для данного недуга. Примерно у 60-80%
пациентов диагностируется дислокация хрусталика из-за
слабости его связочного аппарата, причем еще в
младенческом возрасте. Среди других характерных
признаков:
уплощение роговицы;
увеличение размеров глазного яблока в длину;
миопия или гиперметропия;
нарушение процесса аккомодации из-за недоразвития
цилиарной мышцы.
18. Нервная система
Из-за патологического строения стенок сосудов у
пациентов с СМ повышен риск геморрагических
инсультов, а также кровоизлияний в мозг при разрыве
сосудистых аневризм, субарахноидальных кровотечений.
Среди аномалий развития встречается эктазия твердой
мозговой оболочки. Чаще всего приходится сталкиваться
с пояснично-крестцовой эктазией мозговой оболочки
(выпячивание твердой мозговой оболочки за пределы
позвоночного канала через дефект в строении
позвонков). Это большой критерий СМ, который
встречается в 40% случаев заболевания.
У части пациентов встречаются отклонения в
интеллектуальном развитии, но большинство людей с СМ
характеризируются высокими показателями IQ.
19. Органы системы дыхания
• В большинстве случаев изменения бронхолегочного аппарата
диагностируются случайно. Характерно развитие булл в верхних
частях легких, которые иногда могут разрываться с развитием
спонтанного пневмоторакса.
• Также из-за деформаций грудной клетки пациенты склонны к
развитию эмфиземы легких, частых инфекционных заболеваний
органов дыхания и дыхательной недостаточности.
20. Кожа и мягкие ткани
Встречается повышенная растяжимость кожного покрова,
которая сочетается с развитием атрофических стрий.
Последние появляются спонтанно, они никак не связаны с
колебанием веса, беременностью или гормональными
нарушениями. Подкожный жир выражен слабо у пациентов с
синдромом Марфана. Они часто страдают
рецидивирующими грыжами передней брюшной стенки.
Встречаются и многие другие патологические симптомы
поражения прочих органов и тканей при СМ. Например,
опущение почек (нефроптоз), выпадение мочевого пузыря и
матки у женщин, варикозное расширение вен.
21.
22. Диагностика синдрома Марфана
Диагностика при синдроме Марфана носит в основном клинический
характер. Обязательно учитывают анамнез, в том числе и семейный
(наличие подобных проблем у кого-то из родственников), данные
объективного обследования и осмотра. Также проводят множество
дополнительных диагностических процедур для выявления патологии
тех или иных органов и систем. Для этого применяют ЭКГ, УЗИ сердца и
сосудов, рентгенографию органов грудной клетки, КТ, МРТ внутренних
органов, позвоночника, головного мозга, офтальмоскопию и прочие
исследования органа зрения, аортографию, ангиографию и много других
методик, в зависимости от клинической ситуации и симптомов болезни.
Окончательный диагноз синдрома Марфана выставляют только после
анализа генотипа (ДНК-диагностика) и выявления специфической
мутации в гене, ответственном за продукцию фибриллина, с помощью
молекулярно-генетических методик.
23. Лечение заболевания
К сожалению, на сегодняшний день вылечить синдром
Марфана, как и повлиять на его причину, невозможно. Терапия
в основном направлена на улучшение качества жизни больного
человека, устранение симптомов и профилактику осложнений.
Лечение синдрома Марфана должно быть комплексным и
может включать как консервативные, так и хирургические
методики.
Всем пациентам с СМ рекомендуют ограничения в физической
активности к среднему или низкому уровню, избегать тяжелого
физического труда, не заниматься спортом, так как это
способствует прогрессированию патологии и травматизму.
Больные должны наблюдаться у различных докторов:
кардиолога, окулиста, травматолога-ортопеда, клинического
генетика, невролога
24.
Основная задача в лечении пациентов с Синдромом Марфана заключается в
предотвращении сердечно-сосудистых повреждений
25. Прогноз
Синдром Марфана отличается, как правило,
хроническим прогрессирующим течением.
Продолжительность жизни пациентов при условии
полноценного комплекса лечебных мероприятий в
среднем составляет 45 лет. Основными факторами
риска преждевременной смерти выступают
осложнения, которые возникают вследствие
патологии кардиоваскулярной системы.
26. Синдром Марфана и беременность
Пациентки с СМ могут иметь детей, причем здоровых, но
это очень опасно по двум причинам:
Беременная женщина имеет очень высокий риск летальных
осложнений со стороны сердечно-сосудистой системы. Так
как при вынашивании ребенка создается усиленная
нагрузка на организм матери, а особенно на сердце и
сосуды, то очень большие шансы у пациенток с синдромом
Марфана получить разрыв аневризмы, ее формирование,
расслоение аорты и прочие смертельно опасные
осложнения.
Риск передачи данной наследственной патологии своему
ребенку составляет 50%.
Источник

Презентация по генетике на тему: «Синдром Марфана» Выполнила студентка 24 группы Козлова Анна Оренбург 2014 г.

Синдром Марфана – аутосомно-доминантное генетическое заболевание которое поражает соединительную ткань, характеризующееся диспропорционально длинными конечностями, тонкими худыми пальцами, соответственно худым телосложением и наличием сердечно-сосудистых пороков, которые специфически проявляются в виде пороков сердечных клапанов и аорты.

Синдром марфана – патология соединительной ткани, передающаяся по наследству и приводящая к нарушениям нервной системы, опорнодвигательного аппарата, сердечно – сосудистой системы и других жизненно важных систем организма. Данное заболевание встречается у людей всех рас, в равном соотношении полов.
. В последующие годы эта болезнь наблюдалась")
История Впервые признаки заболевания были описаны Вильямсом (1876). В последующие годы эта болезнь наблюдалась французским профессором педиатрии Антуаном Марфаном, который представил в 1896 году клиническое наблюдение 5 -летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя. Позднее выяснилось, что в действительности девочка страдала врождённой контрактурной арахнодактилией.

Причины возникновения заболевания Причины синдрома марфана окончательно не исследованы. Но считается, что основной дефект связан с нарушением коолагена. Отдельные признаки синдрома Марфана, такие как удлинение пальцев на руках и ногах, заметны уже при рождении ребенка. Но более ярко симптомы синдрома марфана проявляются у детей школьного возраста.

Синдром марфана – довольно редкое генетическое заболевание, его распространенность составляет 1 человек на 5000. Статистика показывает, что в 75% случаях сидром передается от больных родителей детям. В 25% случаях дети с этим синдромом рождаются у абсолютно здоровых родителей. Причины синдрома марфана на сегодня до конца так и не выяснены, генетические мутации возникают в яйцеклетке или сперматозоиде в момент зачатия спонтанно. Дети с синдромом Марфана в вероятности 50% случаях передадут данное заболевание своим детям.

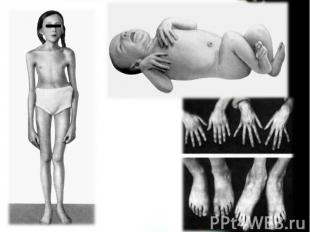
Два брата. Слева нормальный мальчик 10 лет. Справа — 8 -летний мальчик с синдромом Марфана. Высокий рост, отсутствие подкожной жировой клетчатки, сколиоз, деформация грудной клетки

Симптомы Пациенты с синдромом Марфана чаще всего выше остальных родственников и имеют астеническое телосложение, то есть конечности в сравнении с размером туловища гораздо длиннее. Как правило, пальцы на руках и ногах тонкие и длинные. Люди с синдромом Марфана имеют схожие черты лица: они имеют удлиненный череп, глубоко посаженные глаза, неправильный рост зубов и маленькую челюсть.

Со стороны различных органов выделяют следующие симптомы синдрома марфана: Со стороны скелета: высокий рост и длинные конечности могут дополняться искривлением позвоночника и деформацией грудной клетки (грудь становится вдавленной); мягкость суставов; плоскостопие Со стороны глаз: близорукость; «вывих хрусталика» , то есть его смещение с обычного места положения; помутнение хрусталика; отслаивание сетчатки; повышение внутриглазного давления; Со стороны сосудов сердца: Осложнения, связанные с сердцем, являются наиболее серьезными. расширение корня и расслоение стенки аорты. Данные патологии могут привести к внезапному разрыву аорты и вызывать летальный исход; недостаточно плотное закрытие сердечного клапана, приводящее к тому, что кровь течет обратно в сердце. Данная патология приводит к увеличению размеров сердца, шумам в сердце, нерегулярному сердцебиению и отдышке.

Диагностика и профилактика Диагностика синдрома марфана легче всего осуществляется, если у больного или его членов семьи имеется деформация грудной клетки, вывих хрусталика, сильный сколиоз и расширение аорты. Диагностика синдрома марфана осуществляется при мощи таких обследований, как эхокардиография и щелевая лампа.

Лечение синдрома Марфана К сожалению, сегодня не имеется эффективных методик и средств для лечения синдрома Марфана. В основном проводится симптоматическое лечение синдрома Марфана, то есть терапия направлена на снижение риска развития осложнений. При сильном сколиозе назначается проведение физиотерапевтических процедур, также эффективно механическое укрепление скелета. Если угол отклонения позвоночника превышает 45 градусов, показана хирургическая коррекция. Синдром марфана – опасное заболевание, прогноз дальнейшей жизни больного в основном зависит от тяжести изменений со стороны сердечно – сосудистой системы. Разрыв аневризмы аорты и легочного ствола, как правило, приводит к смерти пациента.
Источник
Презентация на тему: Синдром Марфана
Скачать эту презентацию
Скачать эту презентацию
№ слайда 1

Описание слайда:
Синдром Марфана Синдром Марфана
№ слайда 2
, или Марфана-Ашара – это наследственное заболевание соедини")
Описание слайда:
Синдром Марфана (СМ), или Марфана-Ашара – это наследственное заболевание соединительной ткани с преимущественным поражением сердечно-сосудистой системы, скелета и органа зрения. Синдром Марфана (СМ), или Марфана-Ашара – это наследственное заболевание соединительной ткани с преимущественным поражением сердечно-сосудистой системы, скелета и органа зрения. Частота СМ в популяции составляет от 1:3000 до 1:15000. Впервые этот синдром описан французами – в 1896 г., педиатром Антонином Бернардом Марфаном, и в 1902 г. терапевтом Эмилем Шарлем Ашаром.
№ слайда 3

Описание слайда:
Существует интересный факт, что первая девушка модель – Лесли Хорнби, которая послужила прототипом образа всех моделей, имела синдром Марфана. Существует интересный факт, что первая девушка модель – Лесли Хорнби, которая послужила прототипом образа всех моделей, имела синдром Марфана. Как, установлено, что ряд всемирно известных людей страдали синдромом Марфана, среди них следует упомянуть президента США А. Линкольна и великого скрипача Паганини.
№ слайда 4

Описание слайда:
Н. Паганини, Ш. де Голль, Г.Х. Андерсен, Н. Паганини, Ш. де Голль, Г.Х. Андерсен, А. Линкольн.
№ слайда 5

Описание слайда:
СМ относят к наследственным болезням соединительной ткани с аутосомно-доминантным типом наследования. СМ относят к наследственным болезням соединительной ткани с аутосомно-доминантным типом наследования. Молекулярной основой заболевания является нарушение синтеза одного из белков соединительной ткани – фибриллина, который в норме придает ей эластичность и сократимость. При СМ вследствие дефицита фибриллина или его аномального строения соединительная ткань обладает повышенной растяжимостью и теряет способность выдерживать физиологические нагрузки. Ген фибриллина-1 располагается на длинном плече хромосомы 15, и картирован в локусе 15q21. Приблизительно в 75% случаев заболевание передается по наследству, остальные 25% вызываются спорадическими мутациями. Следует сказать, что СМ обладает выраженной генетической гетерогенностью.
№ слайда 6

Описание слайда:
При вступлении в брак 1 больного и 1 здорового родителя вероятность рождения больных детей – 50%. При вступлении в брак 1 больного и 1 здорового родителя вероятность рождения больных детей – 50%.
№ слайда 7

Описание слайда:
В настоящее время в различных семьях идентифицировано более 550 мутаций. В настоящее время в различных семьях идентифицировано более 550 мутаций. Среди обнаруженных мутаций в гене FBN1 57% – миссенс мутации, 18% –фреймшифт (сдвиг рамки считывания), 16% – сплайс сайт, и 8% – нонсенс мутации. Чаще всего при классическом СМ имеет место мутация в одном из доменов FBN1 (epidermal growth factor (EGF)-like domain), ответственных за связывание кальция с фибриллином. Вследствие этого «незащищенный» кальцием фибриллин теряет устойчивость к протеазам, что приводит к дестабилизации микрофибрилл и нарушению их функции. Патологические изменения в одном и том же локусе могут обуславливать разнообразные клинические проявления – от стертой формы с поражением одной из систем организма до классической развернутой.
№ слайда 8

Описание слайда:
I. Форма: I. Форма: 1. Стертая: слабо выраженные изменения в одной, двух системах. 2. Выраженная: а) слабо выраженные изменения в трех системах. б) выраженные изменения хотя бы в одной системе (ограниченная форма). в) выраженные изменения в двух, трех системах и более. II. Характер течения: 1. Прогрессирующий. 2. Стабильный. III. Генетическая характеристика: 1. Семейная форма (тип наследования). 2. Первичная мутация.
№ слайда 9

Описание слайда:
IV. Клинические варианты: IV. Клинические варианты: 1. Болезнь Марфана (присутствие трех классических признаков, семейный характер заболевания). 2. Синдром Марфана (наличие стертых форм с положительными нижеперечисленными диагностическими тестами). 3. Марфаноподобный синдром По МКБ-10 СМ относится к классу XVII: Врожденные аномалии [пороки развития], деформации и хромосомные нарушения; разделу Q87.: Другие уточненные синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем и имеет код Q87.4.
№ слайда 10

Описание слайда:
Поскольку фибриллин находится в соединительной ткани различных органов, симптоматика СМ многосистемна и разнообразна. При этом наиболее часто наблюдается сочетанное поражение сердечно-сосудистой системы, скелета и органа зрения. Поскольку фибриллин находится в соединительной ткани различных органов, симптоматика СМ многосистемна и разнообразна. При этом наиболее часто наблюдается сочетанное поражение сердечно-сосудистой системы, скелета и органа зрения. Естественно, тяжесть состояния и прогноз при СМ зависят, прежде всего, от степени поражения сердца и сосудов. Изменения сердечно-сосудистой системы отмечаются у большинства больных. Их основная причина – потеря способности стенок артерий и клапанных структур сердца выдерживать естественные гемодинамические нагрузки.
№ слайда 11

Описание слайда:
Наиболее частая сердечная патология при СМ – Наиболее частая сердечная патология при СМ – недостаточность митрального клапана. Обычно наблюдается поражение эластических структур створок и сухожильных нитей клапана с развитием его пролабирования и его недостаточности. Эта дисфункция митрального клапана рано или поздно у многих перерастает в умеренную или тяжелую митральную недостаточность, требующую хирургической коррекции. Реже бывает аортальная и трикуспидальная недостаточность. Стенозы клапанов для СМ не характерны. В связи с наличием у больных клапанных пороков, заболевание часто осложняется инфекционным эндокардитом.
№ слайда 12

Описание слайда:
Патологические процессы со стороны аорты при СМ. Расширяется корень аорты, ее клапанное кольцо и синус Вальсальвы. Развивающаяся вследствие этого относительная аортальная недостаточность нередко приводит к кардиомегалии и тяжелой левожелудочковой недостаточности. Патологические процессы со стороны аорты при СМ. Расширяется корень аорты, ее клапанное кольцо и синус Вальсальвы. Развивающаяся вследствие этого относительная аортальная недостаточность нередко приводит к кардиомегалии и тяжелой левожелудочковой недостаточности. Самым грозным осложнением является развитие расслаивающей аневризмы аорты с внутристеночной гематомой, проявляющееся выраженным болевым синдромом и тяжелыми гемодинамическими нарушениями, сто зачастую явлется причиной смерти больных СМ. Идентичные, но менее выраженные изменения могут быть и в легочной артерии. Так как при СМ сосудистая патология генерализованная, поражается эластическая ткань всех сосудов. Аневризмы могут возникать не только в различных отделах аорты, крупных ветвях легочной артерии, но и в венечных, сонных, лучевых, локтевых, бедренных, мозговых и других сосудах.
№ слайда 13

Описание слайда:
Проявления со стороны скелета наблюдаются у 2/3 пациентов. Проявления со стороны скелета наблюдаются у 2/3 пациентов. высокий рост, астеническое телосложение, долихостеномелию, долихоцефалию, прогнатию, "готическое" небо, деформация грудины («куриная» грудь или грудь «сапожника»), арахнодактилию, сколиозы и спондилолистезы, кифосколиозы, нарушение функции суставов, плоскостопие, протрузию вертлужной впадины, дисфункцию височно-нижнечелюстного сустава. Характерным является внешний вид больных: длинные и тонкие конечности с такими же пальцами, длинные, узкие ногти, «птичье лицо» (большой нос и маловыраженный подбородок).
№ слайда 14
№ слайда 15
№ слайда 16

Описание слайда:
– соотношение кисть-рост > 11%; – соотношение кисть-рост > 11%; – отношение размаха рук к росту > 1,05; – длина среднего пальца > 10 см; – отношение длины верхнего сегмента тела к нижнему < 0,86; – индекс телосложения Варги (ИВ) < 1,5 ИВ = масса тела, г/(рост,см)² – возраст, годы/100
№ слайда 17
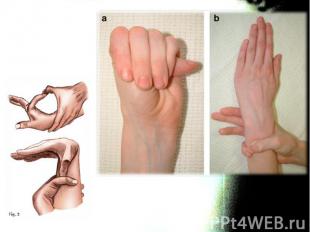
 тест большого пальца Steinberg: согнутый 1-й палец выступает за мягкие ткани")
Описание слайда:
а) тест большого пальца Steinberg: согнутый 1-й палец выступает за мягкие ткани кисти. а) тест большого пальца Steinberg: согнутый 1-й палец выступает за мягкие ткани кисти. При рентгенографии кисти с приведенным большим пальцем его фаланга выступает за скелет метакарпальных костей. б) тест запястья Walker-Murdoch: При обхватывании запястья другой. При арахнодактилии 1-й и 5-й пальцы соединяются друг с другом.
№ слайда 18
№ слайда 19

Описание слайда:
Наиболее часто встречается миопия различной степени, гипоплазия радужки, цилиарной мышцы и пигментной каймы зрачкового края, эктопия хрусталиков кверху, внутрь или кнаружи, реже – изменение калибра сосудов сетчатки, катаракта, зрачковая перепонка, косоглазие, дегенерация сетчатки, врожденная или вторичная глаукома. Наиболее часто встречается миопия различной степени, гипоплазия радужки, цилиарной мышцы и пигментной каймы зрачкового края, эктопия хрусталиков кверху, внутрь или кнаружи, реже – изменение калибра сосудов сетчатки, катаракта, зрачковая перепонка, косоглазие, дегенерация сетчатки, врожденная или вторичная глаукома. Эктопия хрусталиков вследствие надрывов, разрывов и деструкции связок постоянно прогрессирует, что отражается на зрительных функциях и плохо поддается коррекции очками. Наиболее часто эта патология хрусталиков встречается в среднем школьном возрасте, носит двусторонний характер, но степень ее выраженности может быть различной.
№ слайда 20
; Легких: поликистоз, эмфи")
Описание слайда:
Легких: поликистоз, эмфизема, спонтанный пневмоторакс); Легких: поликистоз, эмфизема, спонтанный пневмоторакс); Желудочно-кишечного тракта: висцероптоз, недостаточность кардии; Почек (аплазия, поликистоз). Помимо этого, у больных СМ чаще, чем в общей популяции выявляют рецидивирующие паховые и бедренные грыжи, варикозное расширение вен, разрыв межпозвоночных связок, образования межпозвоночных грыж, опущение мочевого пузыря, матки, атрофические изменения кожи, эктазию твердой мозговой оболочки в пояснично-крестцовом отделе и т.д. Последний симптом считается одним из наиболее важных критериев диагностики заболевания.
№ слайда 21

Описание слайда:
1. Лабораторные. 1. Лабораторные. Наиболее точным лабораторным признаком СМ является генетическая идентификация мутаций в гене FBN1. + показатели почечной экскреции метаболитов соединительной ткани: оксипролина, оксилизилгликозаминов, гликозаминогликанов и их фракционного состава (увел., как повышенный распад коллагена, а его уровень может определять тяжесть заболевания. 2. Электрокардиография. 3. Рентгенография. 4. Компьютерная томография. 5. Ангиография. 6. Эхокардиография. 7. Магнитно-резонансная томография (МРТ) 8. Генеалогический анализ.
№ слайда 22

№ слайда 23

№ слайда 24

№ слайда 25
Описание слайда:
Консервативное. Консервативное. Так как ведущая причина смерти больных СМ – разрыв расслаивающей аневризмы аорты, то консервативное лечение направлено в первую очередь на его предот