
Растяжимость кожи при синдроме элерса данлоса

В этой статье мы расскажем о такой патологии, как синдром Элерса-Данлоса, что это такое, рассмотрим симптомы (фото больных), причины возникновения и метод терапии.
Что такое синдром Элерса-Данлоса
Синдром Элерса-Данлоса – это наследственное заболевание, спровоцированное дефектом синтеза коллагена. При такой заболевании наблюдается неправильное развитие соединительных тканей и гиперэластичность кожи.
Синдром имеет несколько типов, которые могут характеризоваться неестественной растяжимостью кожи, чрезмерной способностью суставов разгибаться в разные стороны, косоглазием, деформацией скелета.
Болезнь затрагивает большое количество систем организма и представляет интерес не только с точки зрения генетики, но и кардиологии, ортопедии, стоматологии, офтальмологии и других медицинских дисциплин.
Так же синдром Элерса-Данлоса отличается сложностью в диагностике из-за наличия легких форм недуга, поэтому нельзя с точностью оценить его распространенность в мире.
Эпидемиология
Точная распространенность и годовая заболеваемость неизвестны, оценки распространенности варьируются от 1/5 до 000-1/20000. Из-за клинической изменчивости эти оценки могут быть слишком низкими. Больше всего болезнь затрагивает – женский пол.
Причины синдрома Элерса-Данлоса
Синдром Элерса-Данлоса имеет несколько вариантов патологии, отличающихся друг от друга по типу наследования. Но их объединяет то , что в их основе лежит структурное либо количественное нарушение белка коллагена. В нынешнее время не для каждого типа заболевания найдены молекулярные механизмы синдрома.
Для первого типа синдрома характерна пониженная активность тех клеток соединительной ткани, отвечающие за синтез межклеточного матрикса. Так же происходит увеличение синтеза протеогликанов, а еще отсутствие ферментов, отвечающих за стабильное воссоздание белков коллагена.
При четвертом типе синдром Элерса-Данлоса наблюдается дефицит коллагена 3 типа. Шестой тип характеризуется недостатком биологического катализатора лизилгидроксилазы, который принимает участие в гидроксилировании лизина в молекулах проколлагена. Седьмой тип заболевания возникает вследствие изменения проколлагена первого типа, который становиться коллагеном.
Если охарактеризовать заболевание в целом, в различных формах, то заболевание возникает вследствие снижения плотности и расстройство ориентации коллагеновых волокон, утончение соединительного отдела кожи, расширение кровеносных сосудов и большую видимость их на поверхности тела.
Симптомы синдрома Элерса-Данлоса
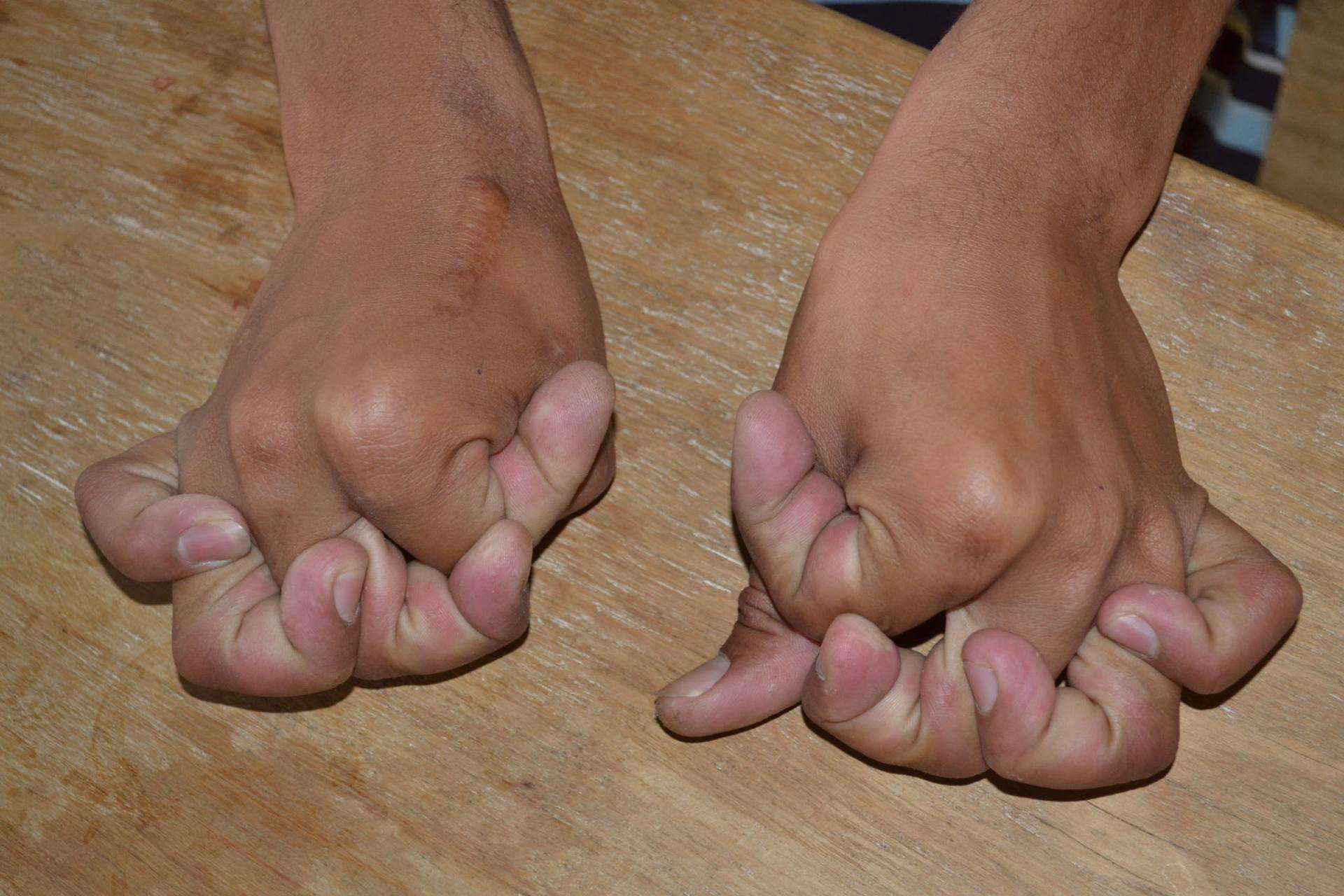
Симптомы могут возникать в любом возрасте, но их трудно диагностировать у маленьких детей из-за повышенной гипервозбудимости суставов. Клинические проявления очень разные. Первичное проявление – это это гиперэластичность различных суставов и аномальная эластичность кожного покрова (см. фото выше).
На ощупь кожа больного нежная, но на ступнях и ладонях присутствуют многочисленные морщины. При синдроме Элерса-Данлоса неестественная эластичность кожи наблюдается с ранних лет жизни человека, так же с возрастом характерно снижение такого отклонения.
Повреждения на коже людей, подверженных синдрому, ощущаются сильней и заживают медленно, а на их месте образуются следы в виде шрамов и опухолей.
Кроме гиперэластичности кожи у подверженных синдрому Элерса-Данлоса наблюдается сильная гиперэластичность суставов, как отдельной части тела, так и всего организма.
Суставы становятся невероятно гибкими и гнуться под неестественными им углами с того момента, как человек начинает учиться ходить, что нередко приводит к травмам. С возрастом ненормальная способность суставов к сгибанию ослабевает.
Повреждение скелета при синдроме представляет собой деформацию грудной клетки в виде дуги или полукруга.
В результате заболевания возникает сколиоз, косолапость. Со скелетом нарушается и расположение внутренних органов, опущение, наличие различных видов грыж, дивертикулез кишечника, пневмоторакс, ввиду нарушения целостности плевры.
Наличие синдрома ведет к патологиям зрения, среди которых встречаются:
- миопия;
- спонтанная отслойка сетчатки;
- косоглазие;
- разрыв глазного яблока и роговицы.
Что касается кардиологии, то у детей с синдромом Элерса-Данлоса возникают частые кровотечения и гематомы.
Не исключен порок сердца, варикоз, пролапс митрального клапана и аневризмы сосудов головного мозга. Нарушений умственного развития детей с данным заболеванием обычно не наблюдается.
Формы синдрома Элерса-Данлоса
Болезнь имеет множество форм:
- Синдром Элерса-Данлоса Тип I (ТИП I ЭДС )
- Синдром Элерса-Данлоса Тип II (ТИП II ЭДС)
- Синдром Элерса-Данлоса III типа (тип ЭДС III и гипермобильности в суставах)
- другие формы (тип IV – тип X)
1 тип синдрома Элерса-Данлоса встречается чаще остальных (40-50%). При таком виде заболевания преобладают симптомы, затрагивающие кожный покров, возможность растяжения которого отходит от нормы в 2-2.5 раз. Данный тип сопровождается наружными кровотечениями и варикозными венами в области нижних конечностей, расшатанностью суставов тела, деформацией скелета. Роды при таком виде заболевания часто происходят преждевременно.
При 2 типе наблюдаются аналогичные признаки, но проявляются они не так сильно. Аномальная гибкость наблюдается только в суставах конечностей, преимущественно стоп и кистей. Растяжимость кожи незначительно отклоняется от нормы, то же и с нарушением работы кровеносных сосудов.
3 тип проявляется ввиду аутосомно-доминантного наследования и протекает доброкачественно. Включает повышенную подвижность суставов по всему телу, деформацию скелета и мышечных тканей и незначительные проявления эластичности кожи.
Редкий и тяжело протекающий 4 тип может быть доминантной и рецессивной. Отличается наличием, у больных повышенной подвижности суставов в пальцах рук, спонтанного возникновения гематом внутренних органов и разрывом всех видов сосудов, что часто приводит к летальным исходам.
5 тип синдрома Элерса-Данлоса происходит из-за X-сцепленного рецессивного наследования. Для него характерны умеренно повышенная мобильность суставов, кровоподтеки и повышенная растяжимость кожи и ее чувствительность.
6 тип наследуется по аутосомно-рецессивному типу. Помимо кровотечений, повышенной подвижности суставов и неестественно эластичной кожи характеризуется увеличением мышечного тонуса (гипертонией мышц), а так же косолапостью и тяжелой формой кифосколиоза, выявляется большой перечень патологий зрения.
7 тип, называемый артроклазией, может наследоваться аутосомно-доминантно и аутосомно-рецессивно. При этом типе у людей часто случаются травмы из-за нездоровой подвижности суставов. Пациентам, подверженным такому типу заболевания, свойственно иметь маленький рост.
8 тип характеризуется аутосомно-доминантным наследованием, и в большей степени для него характерна чувствительность кожи к внешним воздействиям, а так же воспаление тканей, окружающих зубы, что приводит к их ранней потере.
По итогам современных исследований 9 и 10 тип синдрома Элерса-Данлоса не входит в перечень типов классификации заболевания. Для них характерно аутосомно-рецессивное наследование. Кроме гиперэластичности кожи наблюдаются линейные полосы в местах, где кожа наиболее растянута.
Так же присутствует агрегация тромбоцитов и гиперподвижность суставов. У подверженных десятому типу часто наблюдаются врожденные вывихи бедра и постоянно повторяющиеся вывихи плечевых суставов и надколенника.
Диагностика синдрома Элерса-Данлоса
Диагностику синдрома Элерса-Данлоса лучше доверять опытному человеку, специализирующемуся именно в области этого заболевания. Малознакомому с Элерсом-Данлосом врачу будет сложно выявить недуг и тем более его особенности. Это требование связанно с разнообразием индивидуальных проявлений такого заболевания у каждого пациента. Существует множество разновидностей болезни, но есть характерные черты ее проявления.
Неестественная эластичность кожного покрова выявляется путем оттягивания кожи до того момента, пока не почувствуется сопротивление. Участки для осуществления проверки следует выбирать максимально нейтральные, где нормальная оттягиваемость кожи превышает полтора сантиметра.
Среди характерных для болезни синдромов присутствует гипермобильность суставов. Такой симптом определяется по шкале Бейтона, где 5 баллов означает отклонение от нормы. Этот симптом, с возрастом, наблюдается в меньшей степени.
О хрупкости суставных соединений свидетельствуют спонтанно возникающие синяки. Вместе с этим возможны долговременные кровотечения.
Даже при совсем слабых внешних воздействиях появляются различные травмы , которые оставляют атрофические следы ранений в виде шрамов и расширяются с течением времени. С ами ранения заживают дольше, чем у здоровых людей.
Характерной патологией синдрома Элерса-Данлоса является пролапс митрального клапана. Этот недуг выявляется проведением УЗИ сердца.
Постоянная боль в области суставов и конечностей тела – еще одно проявление синдрома Элерса-Данлоса. При таком признаке рентген может не выявить отклонений, а пациент не всегда понимает, из какой части тела исходят болевые ощущения.
Из шкалы Брайтона берутся формальные критерии для того, чтобы диагностировать синдром Элерса-Данлоса. Первоначально функцией шкалы Бейтона была диагностика гипермобильности в суставах, но современные опыты показывают, что синдром гипермобильности суставов и повышенная сгибаемость суставов при Элерсе-Данлосе, – это аналогичные заболевания.
Шкала Бейтона
Основные критерии:
- 4+ баллов по шкале Бейтона выявленных в настоящее время, либо раньше;
- артралгия, длящаяся на протяжении 3 месяцев и более в 4 или более суставах.
Второстепенные критерии:
- 1-3 Балла по шкале Бейтона ( и 0 если вы старше 50-ти);
- артралгия в 1, 2 или 3 суставах с присутствием болезненных ощущений в спине;
- вывихи в одном и более суставах, либо в одном суставе несколько раз;
- более 3 воспалений мягких тканей, ревматизм;
- марфаноидный тип внешности (чрезмерная худощавость, высокий рост, арахнодактилия);
- необычные шрамы и ненормальная растяжимость и тонкость кожи;
- растянутая кожа век или миопия;
- ректальный пролапс, варикозное расширение вен, пролапс матки, грыжа.
Для того чтобы поставить диагноз “синдром Элерса-Данлоса” нужно выявить оба основных критерия, либо 1 из основных и 2 любых второстепенных, для постановки диагноза подойдет и наличие 4 второстепенных критериев.
Когда у вашего первостепенного родственника выявлен данный синдром, вам достаточно будет определить наличие 2 второстепенных критериев.
Лечение синдрома Элерса-Данлоса
Для эффективного устранения синдрома Элерса-Данлоса специфическая терапия до сих пор не разработана, но существуют методы лечения симптомов данной болезни.
Медикаментозная терапия
Для того, чтобы стабилизировать работу сердечно-сосудистой и нервной системы, а так же привести в норму работу суставов, опорно-двигательный аппарат, целостность и эластичность кожного покрова используют несколько вариантов лечения:
- лекарственные препараты в виде аскорбиновой кислоты, А, Е, В витаминов;
- комплексы минералов;
- инъекции соматотропного гормона для стимуляции роста;
- стимулирующие обмен веществ и регенерацию кожного покрова метаболические средства, такие как карнитин-хлорид;
- для стимуляции мозговой деятельности применяются нейрометаболические стимуляторы;
- для поддержания целостности скелета и соединительных тканей используются остеокеа или остеогенон;
- заживлению кожи способствуют такие препараты, как инозин, АТФ, коэнзим Q10.
- используется принимающий участие в синтезе хрящевых и в соединительных тканей глюкозамин.
Хирургическое вмешательство
К хирургическому вмешательству при синдроме Элерса-Данлоса стоит прибегать, только если возникают осложнения, угрожающие жизни. Перед хирургическими операциями должны быть проведены тщательные инвазивные диагностические процедуры для того чтобы оценить степень угрозы и необходимость хирургического вмешательства.
Среди хирургического лечения данного синдрома может быть необходимым: удаление псевдоопухолей, коррекция ВПС, реконструкция грудной стенки и т.д.
Дополнительные и альтернативные методы лечения, в том числе и в домашних условия
Среди процедур назначаются:
- лечебная физкультура и массаж
- рефлексотерапия (воздействие на биологически активные точки организма, отвечающие за работу различных его систем)
- различные физиотерапевтические процедуры (к примеру, лазерная акупунктура)
Также назначается диета с повышенным потреблением белка. Такая диета содержит потребление костных бульонов и заливных блюд.
Прогноз для больных
Повышенного риска ранней смертности нет, однако, в связи с нестабильностью суставов, хроническим и острым болевым синдромом, и симптомами проявляющееся вне костно-мышечной системы, качество жизни больного серьезно ухудшается.
Как уже говорилось выше, специального лечения нет. Необходимо проходить индивидуализированные вспомогательные и симптоматические методы лечения включающие физиотерапию, реабилитацию, прием анальгетиков и соответствующую терапию внесуставных симптомов. Хирургические меры следует рассматривать с умом.
Источник
Синдром Черногубова-Элерса-Данлоса (син. гиперэластическая кожа) представляет собой гетерогенную группу наследственных заболеваний соединительной ткани, характеризующихся рядом общих клинических признаков и сходными морфологическими изменениями. Основные клинические проявления – избыточная пластичность кожи, увеличение подвижности суставов, частые подвывихи, повышенная ранимость кожи, ломкость сосудов с развитием кровоизлияний, гематом при малейших травмах.
К этому синдрому относят 10 типов заболевания, различающихся но наследованию, генетическому дефекту и клинической картине: I – классический тяжелый; II – мягкий; III – доброкачественный гипермобильный; IV – зкхимотический (генный локус 2q31); V – Х-сцепленный рецессивный; VI глазной (генный локус 1р36.3-р36.2); VII – врожденный множественный артрохалазис – генный локус 7q22.10; VIII – с пародонтозом; IX – исключен из классификации синдрома Черногубова-Элерса-Данлоса, обозначается как Х-спепленный вариант вялой кожи; X – дисфибронектинемический; XI – семейная нестабильность суставов. При некоторых формах заболевания предполагается или выявлен первичный биохимический дефект: при I типе – снижение активности фибробластов, усиление синтеза ими протеогликанов, возможно отсутствие ферментов, контролирующих нормальный синтез коллагена; при IV типе – недостаточность продукции III типа коллагена; при VI типе – недостаточность лизилгидроксилазы; при VII типе – патологическое изменение превращения I типа проколлагена в коллаген; при IX типе – недостаточность лизилоксилазы вследствие нарушенного метаболизма меди; при X типе – патологическая функция плазменного фибронектина. Возможно нарушение соотношения гиалуроновая кислота/протеогликаны со значительным повышением содержания гиалуроновой кислоты. Повышенная кровоточивость объясняется изменениями коллагена сосудистой системы и нарушением функционального состояния тромбоцитов.
Патоморфология. Гистологическая картина всех типов синдрома Черногубова-Элерса-Данлоса сходна. Основной гистологический признак – истончение дермы. При этом коллагеновые волокна выглядят нормальными, не теряют своих тинкториальных свойств. Количество эластических волокон относительно увеличено. Число сосудов иногда увеличено и просветы их расширены, вокруг них отмечаются скопления фибробластов и гистиоцитов.
I тип синдрома – классический тяжелый – встречается наиболее часто, на его долю приходится до 43 % всех случаев. Все названные выше признаки заболевания хорошо выражены, но особенно гиперэластичность кожи. Растяжимость кожи увеличена на 100-150 % по сравнению с нормой. Тип наследования аутосомно-доминантный, хотя описаны случаи и рецессивного типа наследования.. Повышенная подвижность суставов носит генерализованный характер, часто развиваются мышечно-скелетные деформации, характерно образование рубцов на месте травм, особенно заметных на лбу, локтях, коленях и лодыжках. Отмечаются сильная ранимость кожи с наклонностью к кровотечениям, плохое заживление ран. Встречаются подкожные опухолевидные элементы, преимущественно в области голеней, моллюскоподобные псевдоопухоли и варикозное расширение вен. У беременных при этом заболевании часты преждевременные роды в результате разрыва плодных оболочек.
Патоморфология. Выражено истончение дермы (примерно наполовину). Размеры пучков коллагеновых волокон неодинаковы, ориентация их нарушена в связи с рыхлостью расположения волокон в пучках, уменьшено их преломление в проходящем свете. При сканирующей электронной микроскопии выявлены нарушение их ориентации, переплетения в виде войлока, потеря компактности строения, утолщения. При трансмиссионной микроскопии обнаружены увеличение среднего диаметра коллагеновых волокон, неравномерность величины и формы фибрилл на поперечных срезах, наличие отдельных гигантских фибрилл, иногда расщепленных на отдельные микрофибриллы. Волокна часто скручены по оси, однако нормальная периодичность сохранена. Отмечаются дистрофические изменения в фибробластах в виде уменьшения их размеров, числа цитоплазматических выростов, слабого развития эндоплазматической сети и вакуолизации цитоплазмы. Подобные изменения коллагеновых волокон вызывают чрезмерную растяжимость кожи. Считают, что нарушение структуры фибрилл происходит на стадии их агрегации и формирования поперечных связей, что может быть обусловлено как нарушением ферментативной регуляции синтеза фибрина, так и изменениями в составе компонентов основного вещества дермы, модулирующих синтез.
II тип синдрома – так называемый мягкий тип, характеризуется теми же признаками, что и тяжелый, но значительно менее выраженными. Растяжимость кожи увеличена только на 30% по сравнению с нормой. Повышение подвижности может отмечаться лишь в суставах кистей и стоп, образование рубцов и наклонность к кровотечениям выражены слабо.
Патоморфология. Толщина дермы близка к норме. При сканирующей электронной микроскопии выявлено уменьшение толщины коллагеновых волокон, а при трансмиссионной – наличие значительного количества коллагеновых волокон с обломанными концами, хотя структура их выглядит нормальной, обнаруживаются единичные фибриллы большого диаметра.
III тип синдрома – доброкачественный гипермобильный, наследуется также аутосомно-доминантно. Основной клинической чертой является повышенная подвижность суставов, носящая генерализованный характер (“человек-змея”), в связи с чем часты ортопедические осложнения и деформации скелета. Гиперэластичность кожи выражена слабо, образование рубцов, как и повышенная ломкость сосудов, выражены минимально.
Патоморфология. Гистологическая картина кожи близка к норме, при электронной микроскопии найдены изменения, сходные с таковыми при I и II типах синдрома, но выраженные в меньшей степени – отсутствуют гигантские коллагеновые волокна и редко находят изменения фибрилл.
Приведенные данные свидетельствуют о близости клинико-морфологических параметров первых трех типов синдрома Черногубова-Элерса-Данло, что позволяет присоединиться к мнению о единой их природе.
IV тип синдрома – экхимотический, наиболее редкий и тяжелый. Установлено, что этот тип генетически неоднороден, описаны как доминантно, так и рецессивно наследуемые варианты. Кожные проявления при всех вариантах аналогичны. Гиперэластичность кожи может быть минимальной. Характерен внешний вид больного: тонкие черты лица, большие глаза, тонкий нос, раннее образование морщин на лице и конечностях (акрогерия). Кожа тонкая и бледная с просвечивающими подкожными сосудами, на ощупь мягкая и бархатистая, на кистях заметно атрофичная. В области костных выступов видны тонкие, пигментированные рубчики, отличающие этот тип синдрома от других. Избыточная подвижность суставов ограничивается пальцами рук. Основным клиническим признаком этого типа является тенденция к кровоточивости. У больных легко возникают экхимозы, нередко обширные при мельчайшей травме, и спонтанно образуются гематомы, особенно на конечностях и во внутренних органах. В некоторых случаях наблюдаются разрывы крупных сосудов, в том числе и аорты. Иногда у больных обнаруживают грыжи пищеварительного тракта, выпадение прямой кишки, спонтанные разрывы полых органов.
Осложненное течение более характерно для рецессивного варианта синдрома, доминантный протекает менее тяжело. В связи с возможностью таких осложнений, как разрывы аорты и полых органов, наступающих, как правило, на третьем десятилетии жизни и приводящих к летальному исходу, необходимы своевременная генетическая консультация и антенатальная диагностика этого заболевания.
Патоморфология. Толщина кожи при IV типе этого синдрома уменьшена на 2/3. При электронно-микроскопическом исследовании обнаружено, что пучки коллагеновых волокон более мелкие, чем в норме, фрагментированные. Толщина коллагеновых фибрилл неравномерная, чаще меньше, чем в норме, отмечено большое количество фибрилл диаметром 60 нм. В основном веществе дермы встречаются скопления мелкогранулярного и волокнистого веществ, протеогликанов. В резко расширенной эндоплазматической сети фибробластов содержатся мелкогранулярные вещества. При исследовании методами электрофоретического и пептидного анализа с использованием расщепления бромцианом коллагена, обнаружено, что в коже больных III тип коллагена содержится в значительно меньших количествах по сравнению с нормой. Поражение кожи и суставов связано главным образом с уменьшением содержания коллагена I типа, в норме преобладающего в них. Своеобразие IV типа синдрома Черногубова-Элерса-Данлоса связано с дефектом коллагена III типа, содержание которого по отношению к коллагену I типа в сосудах и органах пищеварительного тракта значительно выше, чем в коже.
V тип синдрома – Х-сцепленный рецессивный, характеризуется более выраженной гиперэластичностью кожи по сравнению с другими типами, в то время как гипермобильность суставов небольшая. Тенденция к образованию экхимозов и хрупкость кожи выражены умеренно.
Патоморфология. Электронно-микроскопическое исследование кожи выявило сходство изменений с таковыми при I типе синдрома. Биохимически в одном случае выявлен дефект лизиноксидазы – фермента, участвующего в агрегации коллагеновых микрофибрилл и формировании поперечных связей, объединяющих микрофибриллы и коллагеновые фибриллы вне клетки. В других случаях этого дефекта не выявили.
VI тип синдрома – глазной, наследуется по аутосомно-рецессивному типу. При этом типе выражены гиперэластичность кожи, наклонность к кровотечениям, подвижность суставов, отмечается низкий рост больных. Обычно имеются деформации скелета в виде косолапости, тяжелого кифосколиоза, мышечная слабость. Дефект в структуре соединительной ткани глаз приводит к близорукости, кератоконусу, микророговице, глаукоме, отслойке сетчатки, хрупкости склер и роговицы с возможностью их разрыва. Обнаружена недостаточная выработка гидроксилизина и предполагается дефект или мутация лизингидроксилазы – фермента, осуществляющего гидроксилирование лизина во внутриклеточной фазе биосинтеза коллагена, при формировании тройной спирали из полипептидных про-а-цепей. Описано одновременное снижение соотношения коллагена Ш и I типов, что предполагает неоднородность VI типа синдрома.
VII тип синдрома – врожденный множественный артрохалазис, наследуется по аутосомно-рецессивному типу и аутосомно-доминантно. Основное клиническое проявление – гиперподвижность суставов с частыми привычными вывихами, что сближает его с III типом синдрома. В дерме выражено накопление проколлагена. Обнаружен дефект проколлаген пептидазы – фермента, осуществляющего отщепление концевых пептидов протофибрилл, секретируемых фибробластами в процессе образования микрофибрилл.
VIII тип синдрома – с тяжелым пародонтозом, наследуется аутосомно-доминантно, хотя имеется указание и на аутосомно-рецессивный тип наследования. Кожа хрупкая, отмечаются умеренная гипермобильность суставов, слабо выраженная гиперрастяжимость и повышенная кровоточивость кожи, изменения кожи по типу липоидного некробиоза, тяжелый пародонтоз с ранней потерей зубов.
X тип синдрома наследуется аутосомно-рецессивно. Клинически имеются умеренная гиперэластичность и повышенная подвижность суставов, полосовидная атрофия кожи (стрии растяжения). Выявлено нарушение агрегации тромбоцитов, связанное с количественным или качественным дефектом фибронектина, возможно, его a-гранул, содержащихся в тромбоцитах.
XI тип синдрома наследуется аутосомно-доминантно, клинически характеризуется рецидивирующими вывихами суставов, преимущественно плечевых, часты вывихи надколенника, реже наблюдается врожденный вывих бедра. Кожные симптомы выражены слабо. Биохимический дефект заключается в нарушении функции фибронектина плазмы крови.
Гистогенез. В основе клинических проявлений синдрома Черногубова-Элерса-Данлоса лежит нарушение структуры коллагеновых фибрилл. Способность волокон к растяжению связана с образованием ковалентных поперечных связей между микрофибриллами, а также зависит от размеров и целости пучков волокон. Морфологические нарушения проявляются расщеплением отдельных фибрилл, неравномерностью их диаметра и изменением плотности расположения фибрилл в волокнах. Дефект формирования поперечных связей имеется, по-видимому, при всех типах синдрома. Их формирование – конечный этап биосинтеза коллагена, и дефект любого звена биосинтеза может привести к формированию неполноценных волокон. К настоящему времени некоторые дефекты уже известны – дефицит лизиноксидазы при V типе, лизингидроксилазы – при VI, проколлагенпептидазы – при VII. Нарушения метаболизма не всегда связаны с дефектами ферментов биосинтеза коллагена, они могут быть обусловлены факторами микросреды, определенный состав которой обеспечивает нормальный биосинтез.
Проявления синдрома очень многообразны, и не всегда клинически можно определить тип синдрома. Клиническая вариабельность, по-видимому, связана с гетерогенностью коллагена. Так, при IV типе синдрома выявлена недостаточная выработка коллагена III типа, при I-V типах – морфологические изменения коллагена I типа. Биохимическое и морфологическое определение других типов коллагена (в настоящее время выделяют 7 различных его типов)при синдроме Черно-губова-Элерса-Данлоса не проводили.
Источник