
Рото лице пальцевой синдром фото

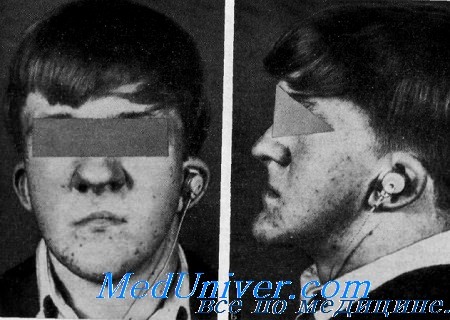
Синдром Мора. Рото-лице-пальцевой или OFD II синдромСиндром, характеризующийся расщеплением губы, дольчатым узловатым языком, широким корнем носа, гипоплазией тела нижней челюсти полидактилией и синдактилией, а также проводящей глухотой, был описан Mohr и Claussen у 4 из 7 сибсов, а также Rimoin и Edgerton у 3 из 4 сибсов. Rimoin и Edgcrton разделили ротолице-пальцевой синдром на две самостоятельные, генетически различные нозологические формы. Одна из них наследуется Х-сцепленным: доминантным путем (OFD I), а другая — аутосомно-рецессивным путем (OFDII). Gorlin, Pindborg и Cohen представили обзор всех опубликованных случаев синдрома. Клинические данные. Данные осмотра. При характеристике лица отмечают значительную гипоплазию скуловых дуг, верхней челюсти и тела нижней челюсти. Переносье широкое и уплощенное. Часто наблюдается срединное расщепление верхней губы, иногда наблюдается расщепление неба и языка. Описаны также жировые гамартомы и общая неподвижность языка. Постоянным симптомом является двусторонняя ульнарная шестипалость на руках и двусторонняя полисиндактилия I пальцев. В немногих случаях отмечается пять пальцев с отклонением V пальца в сторону локтевой кости, синдактилией III и IV пальцев с дополнительными косточками в соединительной ткани пли с шестипалостью только на руках (Gorlin et al.). Орган слуха. Один из 3 сибсов, описанных Rimoin и Edgerton, был мертворожденным; у 2 остальных была выявлена умеренная двусторонняя проводящая глухота. Другие аудиометрические тесты не описаны. У одного из сибсов при помощи тимпанотомии были выявлены врожденное уродство наковальни и недостаточность сустава между наковальней и стременем. Длинный отросток наковальни имел вид «тупой колбасы», а лентовидный отросток вообще отсутствовал. У его сестры также наблюдалась умеренная проводящая глухота. Тимпанотомия не производилась. Goldstein и Medina сообщили, что у обоих сибсов была проводящая глухота с 40% потерей слуха.
Лабораторные данные. Рентгенограммы показали гипоплазию скуловых дуг и тела нижней челюсти. Остальные костные изменения, выявленные главным образом в конечностях (руках и ногах), так же как полидактилия и синдактилия, обсуждены выше. Наследственность. В семье, описанной Mohr и Claussen, были больны 4 брата. Rimoin и Edgerton обнаружили среди 4 сибсов 3 больных. Goldstein и Medina выявили синдром у 2 из 3 сибсов. Некоторые другие примеры выявления синдрома у сибсов опубликовали Gorlin с сотр.. Так как родители во всех случаях были здоровы, аутосомно-рецессивпое наследование очевидно. Диагноз. OFD I синдром характеризуется гипоплазией крыльев носа, преходящей сыпью па лице, тонкими извитыми волосами, нормальным слухом и Х-сцепленным доминантным наследованием с летальностью для мальчиков (Gorlin et al.). Лечение. Расщепление губы, неба и языка может быть восстановлено хирургическим путем. Лишний палец может быть удален. Глухоту также можно лечить хирургическими методами, вставлением протеза вместо аномальных слуховых косточек. Можно также использовать слуховые аппараты. Прогноз. Глухота является врожденной и не прогрессирует. Хотя у большинства больных продолжительность жизни нормальная, некоторые дети умирают от респираторных инфекций (Gorlin et al.). Выводы. Синдром OFD II характеризуется: 1) аутосомно-рецессивным наследованием; 2) аномалиями лица с гипоплазией тела нижней челюсти, уплощением переносья и широко расставленными медиальными углами глаз; 3) аномалиями пальцев, включая полидактилию, синдактилию и брахидактилию; 4) дольчатый язык; 5) проводящую глухоту вследствие уродства слуховых косточек. – Также рекомендуем “Дисплазия эпифиза головки бедра, близорукость и глухота” Оглавление темы “Наследственная глухота”:
|
Источник
Ухо-небно-пальцевой синдром. Ото-палато-дигитальный или OPD-синдромИмеются многочисленные наследственные заболевания, при которых наблюдаются глухота и аномалии костно-мышечной системы. Болезни костной системы распределяются от костных аномалий, охватывающих только несколько костей, таких, как челюстно-лицевой дизостоз и рото-лице-пальцевой синдром II, до генерализованных поражений костной системы, таких, как болезнь Педжета, краниометафизарная дисплазия и склеростеоз. Некоторые из этих синдромов очень редки и встречаются только в одной единственной пораженной семье, например, такие, как глухота и дисгенезия большой берцовой кости. При некоторых синдромах наблюдается проводящая глухота, при других — нейросенсорная или смешанная. OPD-синдром характеризуется проводящей глухотой, задержкой роста, расщеплением неба, лицом «боксера» и общей костной дисплазией. Несколько групп ученых из университета в Миннесоте (Dudding, Gorlin, Langer, Langer, Buran, Duvall) изучили клиническую и рентгенологический) картину синдрома у 3 больных сибсов. Ранее одно наблюдение было описано Taybi. Подробное рентгенологическое исследование большой семьи было проведено Poznanski с соавт.. Клинические данные. Данные осмотра. При рождении ребенок имел нормальную массу и длину, но на протяжении детского возраста его физическое развитие было более замедленным, чем у здоровых детей, масса и рост его были на 10% ниже физиологической нормы. Лицо характеризовалось нависающими бровями с большими надглазничными утолщениями, уплощенной средней частью лица, выступающим затылком, косым антимонголоидным разрезом глаз, гипертелоризмом и широким плоским переносьем. Общий вид больного напоминал боксера. Небо обычно было расщеплено. Могут наблюдаться подвывихи головок лучевой и бедренной костей. Концевые фаланги пальцев широкие, V пальцы короткие с клинодакти-лией; I пальцы на руках были похожи на обрубки, а пальцы на ногах были широкие и кривые, похожие на сучки дерева. Нередко между пальцами имеется перепонка. Орган слуха. Каждый больной считал себя глухим с детского возраста. Аудиометрнческое исследование 3 сибсов показало двустороннюю проводящую глухоту на уровне от 30 до 90 дБ (Buran, Duvall). Двум сибсам была произведена односторонняя тимпанотомия. В каждом случае были обнаружены утолщенные слуховые косточки. У одного сибса был утолщен длинный отросток наковальни и в результате этого сустав между наковальней и стременем был несостоятельным. Головка стремени была расширена, а передняя ножка не достигала основания. У другого сибса ни одна из ножек стремени не достигала основания.
Лабораторные данные. Рентгенограммы. Резко выражены надглазничные бугры. Лобная и затылочная кости утолщены и придают черепу вид гриба. Основание передней черепной ямки также утолщено. Кости лица мелкие, также, как и пазухи верхней челюсти и основной кости. Угол между носовой полостью, турецким седлом и основанием черепа значительно редуцирован. Часто отмечается вывих кзади головки лучевой кости. Па руках видна клинодактилия V пальцев вследствие укорочения средних фаланг со стороны лучевой кости. Дистальные фаланги I—IV пальцев короткие и широкие. Другие аномалии включают добавочный центр окостенения во второй пястной кости и малую многоугольную кость в виде капли слезы. У жепщин-гетерозигот может наблюдаться синостоз между большой многоугольной и ладьевидной костями. Аномалии ног включают боковое искривление бедренных костей и нижней половины большой берцовой кости. Фаланги пальцев на ногах короткие, II и III плюсневые кости в результате слияния с клиновидными костями имеют форму весла; V пястная кость может выступать и содержать дополнительный центр окостенения (Langer, 1967; Gall et al.). Наследственность. Синдром наследуется по Х-сцепленному рецессивному типу. У женщин-гетерозигот наблюдаются выдающиеся боковые части надбровных дуг и малые скелетные аномалии (Gorlin et al., Poznanski et al.). Диагноз. У больных с синдромом Ларсена отмечаются некоторое сходство лица, расщепление неба и вывихи суставов. Однако этот синдром можно отдифференцировать на основании рентгенологических данных (множественные кости запястья, околопяточная кость и т. д.), а также па основании аутосомно-рецессивного или доминантного типа наследования. Лечение. Расщепление неба может быть восстановлено хирургическим путем. Глухоту можно лечить применением слуховых аппаратов или тимпапотомией с введением протезов вместо аномальных слуховых косточек. Выводы. Характеристика этого синдрома включает: 1) Х-сцепленное рецессивное наследование; 2) «лицо боксера», включающее широкий корень носа, гипертелоризм, выступающие лобные и теменные бугры, а также маленькую нижнюю челюсть; 3) расщепление неба; 4) отставание в росте; 5) аномалии рук и ног, включающие широкое расстояние между I и II пальцами и укорочение I пальцев; 6) значительно варьирующие скелетные аномалии; 7) легкую умственную отсталость; 8) умеренную проводящую глухоту. – Также рекомендуем “Синдром Мора. Рото-лице-пальцевой или OFD II синдром” Оглавление темы “Наследственная глухота”:
|
Источник
Блефарофимоз – офтальмологический синдром, проявляющийся двухсторонним птозом в сочетании с уменьшением вертикального и горизонтального размера век. Основные симптомы: патологический разрез глаз, повышенная утомляемость при выполнении зрительной работы, ухудшение зрения, нарушение аккомодации. Для постановки диагноза применяется физикальное обследование, УЗИ глаза, биомикроскопия, визометрия, бесконтактная тонометрия. Тактика лечения сводится к оперативной многоэтапной реконструкции век, включающей кантопластику с последующей коррекцией птоза.
Общие сведения
Блефарофимоз – одна из форм дисплазии век, впервые описанная в 1889 году французским офтальмологом Винем. В 55% случаев наблюдаются спорадические формы патологии. Выявить генетические мутации или установить наследственную предрасположенность при помощи генеалогического метода удается только у 45% больных. Лица мужского и женского пола болеют с одинаковой частотой. Заболевание чаще диагностируют у жителей Северной, Восточной и Юго-Восточной Азии, что обусловлено национальными особенностями внешности. Статистические данные об общей распространенности блефарофимоза в офтальмологии отсутствуют.

Блефарофимоз
Причины блефарофимоза
Этиология болезни до конца не изучена. Заболевание может развиваться изолированно или обнаруживаться в структуре генетических патологий (синдрома Шильбаха-Ротта, рото-лице-пальцевого синдрома). Блефарофимоз часто удается диагностировать у других членов семьи, что говорит о его наследственной природе. Пусковыми факторами являются:
- Тератогенное воздействие. Наиболее выраженное эмбриотоксическое влияние оказывает прием спиртных напитков, курение, потребление наркотических средств во время беременности. Все перечисленное способствует возникновению спорадических мутаций.
- Прием лекарственных средств. Пороки глаз часто вызваны применением беременными медикаментов, которые влияют на формирование глазного яблока. К ним относятся варфарин, антипсихотические средства, наркотические анальгетики, симпатомиметики, стимуляторы нервной системы.
- Инфекционные заболевания. Возникновение симптоматики болезни может быть обусловлено внутриутробным инфицированием вирусом кори, краснухи, герпеса. Вирусные агенты способны проникать через гематоплацентарный барьер и отрицательно воздействовать на эмбриогенез органа зрения.
- Влияние физических факторов. Развитие врожденного блефарофимоза провоцируется радиационным облучением. Обычно это связано с проведением рентгенологической диагностики в период беременности, реже – с воздействием ионизирующего излучения в быту или на производстве.
Патогенез
Блефарофимоз – врожденная, генетически детерминированная патология. Тип наследования – аутосомно-доминантный. Ученые связывают развитие болезни с мутациями гена FOXL2, который локализируется на 3q22.3-q23. Для заболевания характерен феномен образования патологических тканей в зоне орбиты. Формирование спаек приводит к сращению пальпебральных краев, чаще – латерального. Аномалия формируется в первом триместре беременности, когда происходит закладка век и глазного яблока. Воздействие тератогенных факторов на шестом месяце эмбриогенеза плода провоцирует нарушение расщепления фронтоназального и верхнечелюстного отростков на нижнее и верхнее веко.
При внутриутробном заражении вирусами герпеса, краснухи или кори у генетически скомпрометированных лиц нарушается процесс дифференциации структур глазного яблока, это становится причиной множественных пороков развития органа зрения. Синдром блефарофимоза рассматривают как одно из проявлений миогенного, реже нейрогенного птоза. При миогенной природе заболевания дисплазия век сочетается с дистрофическими изменениями круговой мышцы глаза. У лиц с нейрогенной формой наблюдается недоразвитие глазодвигательного нерва. При этом нарушается иннервация кожных покровов в глазничной области.
Симптомы блефарофимоза
Первые симптомы заболевания выявляются в неонатальном периоде. Родители обращают внимание на необычный разрез глаз ребёнка. Глазная щель не только чрезмерно узкая, но и укороченная. Визуализируется деформация медиального угла глаза. У больных определяется недоразвитие переносицы. Расстояние между правым и левым глазным яблоком увеличено, что свидетельствует о гипертелоризме. Также увеличена дистанция между внутренними углами глазных щелей. Обнаруживается вертикальная складка кожи полулунной формы, расположенная в области нижнего века. Верхнее веко опущено. Смыкание глазной щели резко ограничено.
Пациенты старшего возраста предъявляют жалобы на быструю утомляемость глаз при зрительных нагрузках, чрезмерную слезоточивость, чувство инородного тела или «песка» под веками. Из-за узкой глазничной щели нарушено функционирование аккомодационного аппарата, что приводит к зрительной дисфункции. У больных с тяжелым течением заболевания наблюдается отставание в умственном развитии. В случае сопутствующего развития микрофтальма острота зрения снижена вплоть до полной слепоты. У большинства пациентов отмечается повышенная сухость глаз, зачастую выявляется ксерофтальмия. Из-за косметического дефекта нарушается социальная адаптация.
Осложнения
Наиболее распространенное осложнение блефарофимоза – эктропион века. Пациенты подвержены риску инфекционных осложнений переднего сегмента глаз (конъюнктивит, кератит, блефарит). Патология смыкания век и дефектная структура слезной пленки провоцируют возникновение синдрома сухого глаза. Наблюдается высокая вероятность воспаления носослезного канала с последующим развитием дакриоцистита. Дефект костной стенки глазницы ведет к относительному удлинению продольной оси глаза и вторичной гиперметропии. Исключение составляют пациенты с микрофтальмом.
Диагностика
Постановка диагноза базируется на данных физикального обследования и специфических методик. Визуально определяется уменьшение размера вертикальной и горизонтальной глазных щелей, птоз, гипоплазия орбитального края переносицы. Измеряется степень ограничения подвижности верхнего века. Применяются следующие инструментальные методы исследования:
- Визометрия. Характер зрительной дисфункции зависит от степени тяжести заболевания. При легких формах острота зрения соответствует норме. При осложненном течении ухудшение зрения может достигать амавроза.
- Биомикроскопия глаза. При исследовании переднего сегмента глазного яблока выявляется инъекция сосудов конъюнктивы и изъязвления роговой оболочки. Эти изменения носят вторичный характер.
- Бесконтактная тонометрия. В анамнезе у пациентов отмечается нарушение секреции водянистой влаги. В ряде случаев это приводит к снижению внутриглазного давления.
- УЗИ глаза. Ультразвуковое исследование позволяет выявить двухстороннюю симметричную гипоплазию верхних отделов глазницы. Удается визуализировать уменьшение диаметра глазного яблока, что говорит об микрофтальме.
Дифференциальная диагностика проводится с блефарохалазисом и псевдоптозом. При блефарохалазисе развитие клинической симптоматики обусловлено нависанием кожных покровов, расположение верхнего века не изменено. Отличительной чертой псевдоптоза является вторичное опущение верхнего века. Главным этиологическим фактором выступает гипотрофия глазного яблока, эндофтальм, страбизм.
Лечение блефарофимоза
Тактика лечения зависит от анатомических особенностей глазной щели и сопутствующих поражений, разрабатывается индивидуально для каждого пациента. Как правило, больные нуждаются в многоэтапной реконструкции. В первую очередь показана коррекция телекантуса и обратного эпикантуса. Для этого проводят трансназальную фиксацию или Z-пластику. Далее выполняют кантопластику. Цель хирургического вмешательства – коррекция разреза глаз. После проведения кантотомии частично удаляют кантальное сухожилие и фиксируют ткани века к периосту орбиты. В области разреза накладывают косметический шов.
При необходимости на завершающем этапе реконструкции осуществляют коррекцию птоза. При менее выраженном косметическом дефекте ограничиваются проведением кантопексии. В ходе операции подтягивают латеральный пальпебральный уголок. Наружные швы удаляют через 5-7 дней после оперативного вмешательства. Реже дополнительно выполняют пластику медиальной части верхнего века. Пациент должен находиться под динамическим наблюдением у офтальмолога. При необходимости спустя 3-6 месяцев после последней операции проводят корректирующую пластику положения верхнего века.
Прогноз и профилактика
Своевременное проведение оперативного вмешательства позволяет полностью восстановить функции век, устранить или минимизировать косметический дефект. Коррекция зрительной дисфункции при сопутствующем микрофтальме не представляется возможной. Специфическая профилактика блефарофимоза не разработана. Неспецифические превентивные меры базируются на предупреждении воздействия тератогенных факторов (алкоголя, ионизирующего излучения, наркотических средств и пр.) во время вынашивания ребёнка. При наличии случаев блефарофимоза у родителей или близких родственников необходима консультация генетика.
Блефарофимоз – лечение в Москве
Источник