
Рото лице пальцевой синдром презентация

Синдром Мора. Рото-лице-пальцевой или OFD II синдромСиндром, характеризующийся расщеплением губы, дольчатым узловатым языком, широким корнем носа, гипоплазией тела нижней челюсти полидактилией и синдактилией, а также проводящей глухотой, был описан Mohr и Claussen у 4 из 7 сибсов, а также Rimoin и Edgerton у 3 из 4 сибсов. Rimoin и Edgcrton разделили ротолице-пальцевой синдром на две самостоятельные, генетически различные нозологические формы. Одна из них наследуется Х-сцепленным: доминантным путем (OFD I), а другая — аутосомно-рецессивным путем (OFDII). Gorlin, Pindborg и Cohen представили обзор всех опубликованных случаев синдрома. Клинические данные. Данные осмотра. При характеристике лица отмечают значительную гипоплазию скуловых дуг, верхней челюсти и тела нижней челюсти. Переносье широкое и уплощенное. Часто наблюдается срединное расщепление верхней губы, иногда наблюдается расщепление неба и языка. Описаны также жировые гамартомы и общая неподвижность языка. Постоянным симптомом является двусторонняя ульнарная шестипалость на руках и двусторонняя полисиндактилия I пальцев. В немногих случаях отмечается пять пальцев с отклонением V пальца в сторону локтевой кости, синдактилией III и IV пальцев с дополнительными косточками в соединительной ткани пли с шестипалостью только на руках (Gorlin et al.). Орган слуха. Один из 3 сибсов, описанных Rimoin и Edgerton, был мертворожденным; у 2 остальных была выявлена умеренная двусторонняя проводящая глухота. Другие аудиометрические тесты не описаны. У одного из сибсов при помощи тимпанотомии были выявлены врожденное уродство наковальни и недостаточность сустава между наковальней и стременем. Длинный отросток наковальни имел вид «тупой колбасы», а лентовидный отросток вообще отсутствовал. У его сестры также наблюдалась умеренная проводящая глухота. Тимпанотомия не производилась. Goldstein и Medina сообщили, что у обоих сибсов была проводящая глухота с 40% потерей слуха.
Лабораторные данные. Рентгенограммы показали гипоплазию скуловых дуг и тела нижней челюсти. Остальные костные изменения, выявленные главным образом в конечностях (руках и ногах), так же как полидактилия и синдактилия, обсуждены выше. Наследственность. В семье, описанной Mohr и Claussen, были больны 4 брата. Rimoin и Edgerton обнаружили среди 4 сибсов 3 больных. Goldstein и Medina выявили синдром у 2 из 3 сибсов. Некоторые другие примеры выявления синдрома у сибсов опубликовали Gorlin с сотр.. Так как родители во всех случаях были здоровы, аутосомно-рецессивпое наследование очевидно. Диагноз. OFD I синдром характеризуется гипоплазией крыльев носа, преходящей сыпью па лице, тонкими извитыми волосами, нормальным слухом и Х-сцепленным доминантным наследованием с летальностью для мальчиков (Gorlin et al.). Лечение. Расщепление губы, неба и языка может быть восстановлено хирургическим путем. Лишний палец может быть удален. Глухоту также можно лечить хирургическими методами, вставлением протеза вместо аномальных слуховых косточек. Можно также использовать слуховые аппараты. Прогноз. Глухота является врожденной и не прогрессирует. Хотя у большинства больных продолжительность жизни нормальная, некоторые дети умирают от респираторных инфекций (Gorlin et al.). Выводы. Синдром OFD II характеризуется: 1) аутосомно-рецессивным наследованием; 2) аномалиями лица с гипоплазией тела нижней челюсти, уплощением переносья и широко расставленными медиальными углами глаз; 3) аномалиями пальцев, включая полидактилию, синдактилию и брахидактилию; 4) дольчатый язык; 5) проводящую глухоту вследствие уродства слуховых косточек. – Также рекомендуем “Дисплазия эпифиза головки бедра, близорукость и глухота” Оглавление темы “Наследственная глухота”:
|
Источник
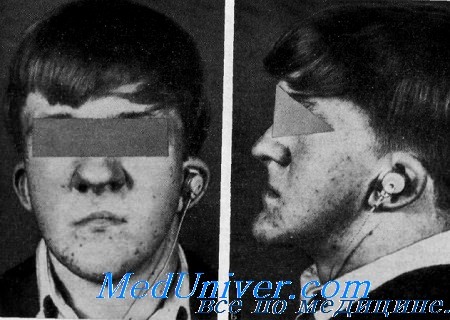
Ухо-небно-пальцевой синдром. Ото-палато-дигитальный или OPD-синдромИмеются многочисленные наследственные заболевания, при которых наблюдаются глухота и аномалии костно-мышечной системы. Болезни костной системы распределяются от костных аномалий, охватывающих только несколько костей, таких, как челюстно-лицевой дизостоз и рото-лице-пальцевой синдром II, до генерализованных поражений костной системы, таких, как болезнь Педжета, краниометафизарная дисплазия и склеростеоз. Некоторые из этих синдромов очень редки и встречаются только в одной единственной пораженной семье, например, такие, как глухота и дисгенезия большой берцовой кости. При некоторых синдромах наблюдается проводящая глухота, при других — нейросенсорная или смешанная. OPD-синдром характеризуется проводящей глухотой, задержкой роста, расщеплением неба, лицом «боксера» и общей костной дисплазией. Несколько групп ученых из университета в Миннесоте (Dudding, Gorlin, Langer, Langer, Buran, Duvall) изучили клиническую и рентгенологический) картину синдрома у 3 больных сибсов. Ранее одно наблюдение было описано Taybi. Подробное рентгенологическое исследование большой семьи было проведено Poznanski с соавт.. Клинические данные. Данные осмотра. При рождении ребенок имел нормальную массу и длину, но на протяжении детского возраста его физическое развитие было более замедленным, чем у здоровых детей, масса и рост его были на 10% ниже физиологической нормы. Лицо характеризовалось нависающими бровями с большими надглазничными утолщениями, уплощенной средней частью лица, выступающим затылком, косым антимонголоидным разрезом глаз, гипертелоризмом и широким плоским переносьем. Общий вид больного напоминал боксера. Небо обычно было расщеплено. Могут наблюдаться подвывихи головок лучевой и бедренной костей. Концевые фаланги пальцев широкие, V пальцы короткие с клинодакти-лией; I пальцы на руках были похожи на обрубки, а пальцы на ногах были широкие и кривые, похожие на сучки дерева. Нередко между пальцами имеется перепонка. Орган слуха. Каждый больной считал себя глухим с детского возраста. Аудиометрнческое исследование 3 сибсов показало двустороннюю проводящую глухоту на уровне от 30 до 90 дБ (Buran, Duvall). Двум сибсам была произведена односторонняя тимпанотомия. В каждом случае были обнаружены утолщенные слуховые косточки. У одного сибса был утолщен длинный отросток наковальни и в результате этого сустав между наковальней и стременем был несостоятельным. Головка стремени была расширена, а передняя ножка не достигала основания. У другого сибса ни одна из ножек стремени не достигала основания.
Лабораторные данные. Рентгенограммы. Резко выражены надглазничные бугры. Лобная и затылочная кости утолщены и придают черепу вид гриба. Основание передней черепной ямки также утолщено. Кости лица мелкие, также, как и пазухи верхней челюсти и основной кости. Угол между носовой полостью, турецким седлом и основанием черепа значительно редуцирован. Часто отмечается вывих кзади головки лучевой кости. Па руках видна клинодактилия V пальцев вследствие укорочения средних фаланг со стороны лучевой кости. Дистальные фаланги I—IV пальцев короткие и широкие. Другие аномалии включают добавочный центр окостенения во второй пястной кости и малую многоугольную кость в виде капли слезы. У жепщин-гетерозигот может наблюдаться синостоз между большой многоугольной и ладьевидной костями. Аномалии ног включают боковое искривление бедренных костей и нижней половины большой берцовой кости. Фаланги пальцев на ногах короткие, II и III плюсневые кости в результате слияния с клиновидными костями имеют форму весла; V пястная кость может выступать и содержать дополнительный центр окостенения (Langer, 1967; Gall et al.). Наследственность. Синдром наследуется по Х-сцепленному рецессивному типу. У женщин-гетерозигот наблюдаются выдающиеся боковые части надбровных дуг и малые скелетные аномалии (Gorlin et al., Poznanski et al.). Диагноз. У больных с синдромом Ларсена отмечаются некоторое сходство лица, расщепление неба и вывихи суставов. Однако этот синдром можно отдифференцировать на основании рентгенологических данных (множественные кости запястья, околопяточная кость и т. д.), а также па основании аутосомно-рецессивного или доминантного типа наследования. Лечение. Расщепление неба может быть восстановлено хирургическим путем. Глухоту можно лечить применением слуховых аппаратов или тимпапотомией с введением протезов вместо аномальных слуховых косточек. Выводы. Характеристика этого синдрома включает: 1) Х-сцепленное рецессивное наследование; 2) «лицо боксера», включающее широкий корень носа, гипертелоризм, выступающие лобные и теменные бугры, а также маленькую нижнюю челюсть; 3) расщепление неба; 4) отставание в росте; 5) аномалии рук и ног, включающие широкое расстояние между I и II пальцами и укорочение I пальцев; 6) значительно варьирующие скелетные аномалии; 7) легкую умственную отсталость; 8) умеренную проводящую глухоту. – Также рекомендуем “Синдром Мора. Рото-лице-пальцевой или OFD II синдром” Оглавление темы “Наследственная глухота”:
|
Источник
1.
Наследственность и патология
2.
НАСЛЕДСТВЕННОСТЬ
свойство организмов
сохранять и обеспечивать передачу
морфофункциональных признаков
потомкам;
программировать особенности
их индивидуального развития
в конкретных условиях среды.
3.
ИЗМЕНЧИВОСТЬ
свойство организмов
приобретать в онтогенезе новые
морфо-функциональные признаки
и особенности индивидуального
развития,
отличающиеся от родительских.
4.
ВИДЫ ИЗМЕНЧИВОСТИ
ФЕНОТИПИЧЕСКАЯ
ГЕНОТИПИЧЕСКАЯ
(ненаследуемая,
модификационная)
(наследуемая)
ФЕНОКОПИИ
СОМАТИЧЕСКАЯ
ГЕНЕРАТИВНАЯ
МУТАЦИОННАЯ
КОМБИНАТИВНАЯ
5.
ВИДЫ МУТАЦИЙ
ПО ПРИЧИНЕ
“СПОНТАННЫЕ”
ИНДУЦИРОВАННЫЕ
ПО ВИДУ КЛЕТОК,
В КОТОРЫХ
ПРОИЗОШЛА МУТАЦИЯ
СОМАТИЧЕСКИЕ
ГАМЕТИЧЕСКИЕ
ПО ЗНАЧЕНИЮ
ПАТОГЕННЫЕ
НЕЙТРАЛЬНЫЕ
БЛАГОПРИЯТНЫЕ
ПО “УРОВНЮ”
(“МАСШТАБУ”)
ГЕННЫЕ
ХРОМОСОМНЫЕ
ГЕНОМНЫЕ
6.
ВИДЫ МУТАЦИЙ “ПО МАСШТАБУ”
ГЕННЫЕ
изменения ДНК
ХРОМОСОМНЫЕ
ГЕНОМНЫЕ
изменения структуры
отдельных хромосом
изменения
числа хромосом
П Р И М Е Р Ы:
* гемоглобиноз S
* гемофилии
* муковисцидоз
* нейрофиброматоз
* фенилкетонурия
* делеция хромосом
(5р – синдром
“кошачьего крика”)
* дупликация
короткого плеча
хромосомы 9
(множественные ВПР)
* полиплоидии
* анеуплоидии
(моносомии,
трисомии)
7.
8.
ВИДЫ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
ГЕННЫЕ
ХРОМОСОМНЫЕ
БОЛЕЗНИ
БОЛЕЗНИ
СОБСТВЕННО
ХРОМОСОМНЫЕ
ГЕНОМНЫЕ
БОЛЕЗНИ
ГЕНЕТИЧЕСКОЙ
НЕСОВМЕСТИМОСТИ
МАТЕРИ И ПЛОДА
ГЕНЕТИЧЕСКИЕ
СОМАТИЧЕСКИЕ
БОЛЕЗНИ
БОЛЕЗНИ
С ГЕНЕТИЧЕСКОЙ
ПРЕДРАСПОЛОЖЕННОСТЬЮ
(МНОГОФАКТОРНЫЕ)
9.
ВИДЫ ГЕННЫХ БОЛЕЗНЕЙ
В ЗАВИСИМОСТИ ОТ ТИПА ИХ НАСЛЕДОВАНИЯ (1)
АУТОСОМНО-
АУТОСОМНО-
ДОМИНАНТНЫЕ
РЕЦЕССИВНЫЕ
ДОМИНАНТНЫЕ,
СЦЕПЛЕННЫЕ
С
Х
ХРОМОСОМОЙ
РЕЦЕССИВНЫЕ,
СЦЕПЛЕННЫЕ
С
Х
ХРОМОСОМОЙ
ПРИМЕРЫ
*ПОЛИДАКТИЛИЯ
*СИНДРОМ МАРФАНА
*ГИПЕРХОЛЕСТЕРИНЕМИЯ СЕМЕЙНАЯ
*НЕЙРОФИБРОМАТОЗ
*М-ГЕМОГЛОБИНОЗ *ХОРЕЯ
ГЕНТИНГТОНА
*ПОЛИПОЗ ТОЛСТОГО
КИШЕЧНИКА
*ГАЛАКТОЗЕМИЯ
*ФЕНИЛКЕТОНУРИЯ
*S- ГЕМОГЛОБИНОЗ
*АЛЬБИНИЗМ
*ГЛИКОГЕНОЗЫ
*МУКОВИСЦИДОЗ
*АДРЕНОГЕНИТАЛЬНЫЙ
СИНДРОМ
*ГИПЕРЛИПОПРОТЕИНЕМИЯ
*РАХИТ,УСТОЙЧИВЫЙ
К ВИТАМИНУ D
*РОТО-ЛИЦЕ-ПАЛЬЦЕВОЙ
СИНДРОМ
*ФРОНТОНАЗАЛЬНАЯ
ДИСПЛАЗИЯ
*КАТАРАКТА
*ГЕМОФИЛИИ А, В
*ДАЛЬТОНИЗМ
*ГИПОГАМОГЛОБУЛИНЕМИЯ
*МЫШЕЧНАЯ
ДИСТРОФИЯ ДЮШЕННА
10.
ВИДЫ ГЕННЫХ БОЛЕЗНЕЙ
В ЗАВИСИМОСТИ ОТ ТИПА ИХ НАСЛЕДОВАНИЯ (2)
ГОЛАНДРИЧЕСКИЕ
МИТОХОНДРИАЛЬНЫЕ
ПРИМЕРЫ
* ИЗБЫТОЧНОЕ ОВОЛОСЕНИЕ
УШНЫХ РАКОВИН
* АЗООСПЕРМИЯ
*АТРОФИЯ ЗРИТЕЛЬНОГО НЕРВА
ЛЕБЕРА
*ЭНЦЕФАЛОПАТИЯ
МИТОХОНДРИАЛЬНАЯ
*ЭПИЛЕПСИЯ МИОКЛОНАЛЬНАЯ
*КАРДИОМИОПАТИЯ
11.
ВИДЫ ХРОМОСОМНЫХ МУТАЦИЙ
ВНУТРИХРОМОСОМНЫЕ
МЕЖХРОМОСОМНЫЕ
РЕЦИПРОКНЫЕ
ДЕЛЕЦИИ
ИНВЕРСИИ
ДУПЛИКАЦИИ
НЕРЕЦИПРОКНЫЕ
“ЦЕНТРИЧЕСКОЕ”
СЛИЯНИЕ
12.
ВИДЫ ХРОМОСОМНЫХ БОЛЕЗНЕЙ
В ЗАВИСИМОСТИ ОТ НАРУШЕНИЯ
СТРУКТУРЫ ИЛИ ЧИСЛА ХРОМОСОМ
НАРУШЕНИЕ
СТРУКТУРЫ
ХРОМОСОМ
ИЗМЕНЕНИЕ
ЧИСЛА
ХРОМОСОМ
ИЛИ
ПЛОИДНОСТИ
В ЗАВИСИМОСТИ ОТ ВИДА КЛЕТОК,
В КОТОРЫХ ПРОИЗОШЛА МУТАЦИЯ
ПОЛНЫЕ
ФОРМЫ
С ИЗМЕНЕНИЕМ
ЧИСЛА
ХРОМОСОМ
СТРУКТУРЫ
ХРОМОСОМ
МОЗАИЧНЫЕ
ФОРМЫ
С МУТАЦИЯМИ
ХРОМОСОМНЫМИ
ГЕНОМНЫМИ
13.
ВИДЫ ГЕНОМНЫХ МУТАЦИЙ
ПОЛИПЛОИДИЯ
(3n, 4n, …)
АНЭУПЛОИДИЯ
(2n ± 1)
14.
ВИДЫ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
ПО ВИДУ МУТАНТНЫХ
КЛЕТОК
ГАМЕТИЧЕСКИЕ
СОМАТИЧЕСКИЕ
ПО РОЛИ ФАКТОРОВ
НАСЛЕДСТВЕННОСТИ И СРЕДЫ
В ИХ ВОЗНИКНОВЕНИИ
СОБСТВЕННО
НАСЛЕДСТВЕННЫЕ
ВОЗНИКАЮЩИЕ
ПРИ ДЕЙСТВИИ
ОПРЕДЕЛЕННОГО
ФАКТОРА СРЕДЫ
ВЫЗЫВАЕМЫЕ
ФАКТОРАМИ
СРЕДЫ
КОМБИНИРОВАННЫЕ
ВОЗНИКАЮЩИЕ ПРИ ДЕЙСТВИИ
ОПРЕДЕЛЕННОГО ФАКТОРА СРЕДЫ НА
“ПРЕДРАСПОЛОЖЕННЫЙ” ОРГАНИЗМ
15.
СИНДРОМ
МАРФАНА
ПРОЯВЛЕНИЯ
ПОРАЖЕНИЕ
СКЕЛЕТА
ПОРАЖЕНИЕ
СЕРДЦА И СОСУДОВ
* длинные конечности
* пролапс
митрального клапана
* арахнодактилия
* аневризма аорты
* высокий рост
* гиперподвижность
суставов
ПОРАЖЕНИЕ
ГЛАЗ
* вывих хрусталика
* дрожание радужки
16.
Возможные наборы половых хромосом
при нормальном и аномальном течении
I мейотического деления
яйцеклетки
спермии
Х
Y
Х
О
ХХ
ХХ
ХО
ХХХ
норма
летальный
полисомия Х
ХY
YО
с-м Клайнфельтера
норма
летальный
О
ХО
с-м Шерешевского-
ОО
Тернера
летальный
ХY
с-м Клайнфельтера
ХХY
ХY
норма?
ХХY
ХХ
норма?
ХХХY
с-м Клайнфельтера
17.
Врождённый порок развития (ВПР)
* Стойкое морфологическое изменение органа, его
части или участка тела,
* выходящее за пределы
нормального диапазона строения.
*Возникает внутриутробно.
* Обусловливает расстройство
жизнедеятельности организма.
18.
НАИБОЛЕЕ ЧАСТЫЕ
ВРОЖДЕННЫЕ ПОРОКИ РАЗВИТИЯ
АГЕНЕЗИЯ
АТРЕЗИЯ
ГЕТЕРОТОПИЯ
АПЛАЗИЯ,
ГИПОПЛАЗИЯ
УДВОЕНИЕ/
УТРОЕНИЕ
СТЕНОЗ
ПЕРСИСТИРОВАНИЕ
ЭКТОПИЯ
19.
ВИДЫ ВРОЖДЕННЫХ ПОРОКОВ РАЗВИТИЯ
В ЗАВИСИМОСТИ ОТ ОБЪЕКТА ВОЗДЕЙСТВИЯ
ПОВРЕЖДАЮЩИХ ФАКТОРОВ И СРОКА БЕРЕМЕННОСТИ
ГАМЕТОПАТИИ
*объект: половые
клетки
БЛАСТОПАТИИ
*объект: бластоциты
*срок: первые 15 суток
после оплодотворения
ЭМБРИОПАТИИ
*объект: эмбрион
*срок: 16 день –
8-9 неделя
беременности
ФЁТОПАТИИ
*объект: плод
*срок: после 8-9
недели
беременности
20.
СВОЙСТВА МУЛЬТИФАКТОРИАЛЬНЫХ ЗАБОЛЕВАНИЙ
в этиологии
важна
роль изменений
в геноме
предрасположенность
к болезни зависит
от большого числа генов
(феномен аддитивности)
характер наследования
не объясняется
только менделевскими
законами
предрасположенность
реализуется под влиянием
большого числа
факторов среды
П р и м е р ы:
* Ишемическая болезнь сердца (ИБС)
* Гипертоническая болезнь
* Бронхиальная астма
* Сахарный диабет
* Язвенная болезнь желудка и кишечника
* Псориаз
* Эпилепсия
* Системная красная волчанка
*…
21.
ОСНОВНЫЕ МЕТОДЫ ДИАГНОСТИКИ И АНАЛИЗА
ПАТОГЕНЕЗА НАСЛЕДСТВЕННЫХ ФОРМ ПАТОЛОГИИ
клиникосиндромологический
клиникогенеалогический
генетики
соматических
клеток
клонирования
селекции
гибридизации
цитогенетический
биологического
моделирования
близнецовый
биохимический
молекулярногенетический
гибридизация ДНК
клонирование ДНК
полимеразная цепная
реакция
блотгибридизации ДНК
22.
ПРИНЦИПЫ И МЕТОДЫ ЛЕЧЕНИЯ
НАСЛЕДСТВЕННЫХ ФОРМ ПАТОЛОГИИ
ПРИНЦИПЫ
МЕТОДЫ
ЭТИОТРОПНЫЙ
* коррекция дефекта
генома:
– введение в геном
нормального гена,
– подавление
репликации
патогенного гена
* изменение генома:
– введение в него гена,
кодирующего синтез
чужеродного для
системы ИБН
антигена
ПАТОГЕНЕТИЧЕСКИЙ
* заместительная терапия
(введение “дефицитного”
вещества)
СИМПТОМАТИЧЕСКИЙ
* устранение тягостных,
усугубляющих
состояние симптомов
* коррекция метаболизма:
– ограничения попадания в
организм неметаболизируемых
веществ (лактозы, фенилаланина),
– выведение избытка метаболитов
(холестерина, фенилпировиноградной
кислоты),
– регуляция активности ферментов
(липопротеинлипазы крови, КФКазы)
* хирургическое устранение дефектов
(шунтов, сращений, создание шунтов)
Источник
1
Наследственность и патология © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
2
Н А С Л Е Д С Т В Е Н Н О С Т Ь * свойство организмов * сохранять и обеспечивать передачу морфо-функциональных признаков потомкам; * программировать особенности их индивидуального развития в конкретных условиях среды. © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
3
И З М Е Н Ч И В О С Т Ь * свойство организмов * приобретать в онтогенезе новые морфо-функциональные признаки и особенности индивидуального развития, * отличающиеся от родительских. © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
4
ВИДЫ ИЗМЕНЧИВОСТИ ГЕНОТИПИЧЕСКАЯ (наследуемая) СОМАТИЧЕСКАЯГЕНЕРАТИВНАЯ КОМБИНАТИВНАЯ ФЕНОТИПИЧЕСКАЯ (ненаследуемая, модификационная) ФЕНОКОПИИ МУТАЦИОННАЯ © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
5
ВИДЫ МУТАЦИЙ ПО ЗНАЧЕНИЮ ПО ВИДУ КЛЕТОК, В КОТОРЫХ ПРОИЗОШЛА МУТАЦИЯ СОМАТИЧЕСКИЕ СПОНТАННЫЕ ИНДУЦИРОВАННЫЕ ПО ПРИЧИНЕ ПО УРОВНЮ (МАСШТАБУ) ГАМЕТИЧЕСКИЕ ГЕННЫЕ ПАТОГЕННЫЕ ХРОМОСОМНЫЕ НЕЙТРАЛЬНЫЕ БЛАГОПРИЯТНЫЕ ГЕНОМНЫЕ © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
6
ВИДЫ МУТАЦИЙ ПО МАСШТАБУ * изменения ДНК * изменения структуры отдельных хромосом * изменения числа хромосом П Р И М Е Р Ы * гемоглобиноз S * гемофилии * муковисцидоз * нейрофиброматоз * фенилкетонурия * делеция хромосом (5р – синдром кошачьего крика) * дупликация короткого плеча хромосомы 9 (множественные ВПР) * полиплоидии * анеуплоидии (моносомии, трисомии) ГЕННЫЕХРОМОСОМНЫЕГЕНОМНЫЕ © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
7
ОСНОВНЫЕ МЕХАНИЗМЫ ГЕННЫХ МУТАЦИЙ ДУПЛИКАЦИЯ УЧАСТКА ДНК ИНВЕРСИЯ СЕГМЕНТА ДНК ИНСЕРЦИЯ ФРАГМЕНТА ДНК ТРАНСВЕРСИЯ ОСНОВАНИЙ ТРАНЗИЦИЯ ОСНОВАНИЙ ДЕЛЕЦИЯ СЕГМЕНТА ДНК © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
8
ВИДЫ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ БОЛЕЗНИ С ГЕНЕТИЧЕСКОЙ ПРЕДРАСПОЛОЖЕННОСТЬЮ (МНОГОФАКТОРНЫЕ) БОЛЕЗНИ ГЕНЕТИЧЕСКОЙ НЕСОВМЕСТИМОСТИ МАТЕРИ И ПЛОДА СОБСТВЕННО ХРОМОСОМНЫЕ ГЕНОМНЫЕ ГЕННЫЕ БОЛЕЗНИ ХРОМОСОМНЫЕ БОЛЕЗНИ ГЕНЕТИЧЕСКИЕ СОМАТИЧЕСКИЕ БОЛЕЗНИ © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
9
ВИДЫ ГЕННЫХ БОЛЕЗНЕЙ В ЗАВИСИМОСТИ ОТ ТИПА ИХ НАСЛЕДОВАНИЯ (1) ВИДЫ ГЕННЫХ БОЛЕЗНЕЙ В ЗАВИСИМОСТИ ОТ ТИПА ИХ НАСЛЕДОВАНИЯ (1) РЕЦЕССИВНЫЕ, СЦЕПЛЕННЫЕ С Х ХРОМОСОМОЙ АУТОСОМНО- ДОМИНАНТНЫЕ ДОМИНАНТНЫЕ, СЦЕПЛЕННЫЕ С Х ХРОМОСОМОЙ *ПОЛИДАКТИЛИЯ *СИНДРОМ МАРФАНА *ГИПЕРХОЛЕСТЕРИН- ЕМИЯ СЕМЕЙНАЯ *НЕЙРОФИБРОМАТОЗ *М-ГЕМОГЛОБИНОЗ *ХОРЕЯ ГЕНТИНГТОНА *ПОЛИПОЗ ТОЛСТОГО КИШЕЧНИКА *ГАЛАКТОЗЕМИЯ *ФЕНИЛКЕТОНУРИЯ *S- ГЕМОГЛОБИНОЗ *АЛЬБИНИЗМ *ГЛИКОГЕНОЗЫ *МУКОВИСЦИДОЗ *АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ *ГИПЕРЛИПОПРОТЕИНЕМИЯ *РАХИТ,УСТОЙЧИВЫЙ К ВИТАМИНУ D *РОТО-ЛИЦЕ-ПАЛЬЦЕВОЙ СИНДРОМ *ФРОНТОНАЗАЛЬНАЯ ДИСПЛАЗИЯ *КАТАРАКТА *ГЕМОФИЛИИ А, В *ДАЛЬТОНИЗМ *ГИПОГАМО- ГЛОБУЛИНЕМИЯ *МЫШЕЧНАЯ ДИСТРОФИЯ ДЮШЕННА П Р И М Е Р Ы АУТОСОМНО- РЕЦЕССИВНЫЕ © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
10
ВИДЫ ГЕННЫХ БОЛЕЗНЕЙ В ЗАВИСИМОСТИ ОТ ТИПА ИХ НАСЛЕДОВАНИЯ (2) ВИДЫ ГЕННЫХ БОЛЕЗНЕЙ В ЗАВИСИМОСТИ ОТ ТИПА ИХ НАСЛЕДОВАНИЯ (2) * ИЗБЫТОЧНОЕ ОВОЛОСЕНИЕ УШНЫХ РАКОВИН * АЗООСПЕРМИЯ *АТРОФИЯ ЗРИТЕЛЬНОГО НЕРВА ЛЕБЕРА *ЭНЦЕФАЛОПАТИЯ МИТОХОНДРИАЛЬНАЯ *ЭПИЛЕПСИЯ МИОКЛОНАЛЬНАЯ *КАРДИОМИОПАТИЯ ГОЛАНДРИЧЕСКИЕМИТОХОНДРИАЛЬНЫЕ П Р И М Е Р Ы © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
11
ВИДЫ ХРОМОСОМНЫХ МУТАЦИЙ НЕРЕЦИПРОКНЫЕ ВНУТРИХРОМОСОМНЫЕ МЕЖХРОМОСОМНЫЕ ИНВЕРСИИ ДЕЛЕЦИИ РЕЦИПРОКНЫЕ ЦЕНТРИЧЕСКОЕ СЛИЯНИЕ ДУПЛИКАЦИИ © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
12
В ЗАВИСИМОСТИ ОТ НАРУШЕНИЯ СТРУКТУРЫ ИЛИ ЧИСЛА ХРОМОСОМ В ЗАВИСИМОСТИ ОТ ВИДА КЛЕТОК, В КОТОРЫХ ПРОИЗОШЛА МУТАЦИЯ НАРУШЕНИЕ СТРУКТУРЫ ХРОМОСОМ ИЗМЕНЕНИЕ ЧИСЛА ХРОМОСОМ ИЛИ ПЛОИДНОСТИ ПОЛНЫЕ ФОРМЫ С ИЗМЕНЕНИЕМ ВИДЫ ХРОМОСОМНЫХ БОЛЕЗНЕЙ ЧИСЛА ХРОМОСОМ СТРУКТУРЫ ХРОМОСОМ ХРОМОСОМНЫМИ ГЕНОМНЫМИ МОЗАИЧНЫЕ ФОРМЫ С МУТАЦИЯМИ © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
13
ВИДЫ ГЕНОМНЫХ МУТАЦИЙ АНЭУПЛОИДИЯ (2n ± 1) ПОЛИПЛОИДИЯ (3n, 4n, …) © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
14
СОМАТИЧЕСКИЕГАМЕТИЧЕСКИЕ ВОЗНИКАЮЩИЕ ПРИ ДЕЙСТВИИ ОПРЕДЕЛЕННОГО ФАКТОРА СРЕДЫ НА ПРЕДРАСПОЛОЖЕННЫЙ ОРГАНИЗМ ВЫЗЫВАЕМЫЕ ФАКТОРАМИ СРЕДЫ ВОЗНИКАЮЩИЕ ПРИ ДЕЙСТВИИ ОПРЕДЕЛЕННОГО ФАКТОРА СРЕДЫ СОБСТВЕННО НАСЛЕДСТВЕННЫЕ ВИДЫ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ ПО ВИДУ МУТАНТНЫХ КЛЕТОК ПО РОЛИ ФАКТОРОВ НАСЛЕДСТВЕННОСТИ И СРЕДЫ В ИХ ВОЗНИКНОВЕНИИ КОМБИНИРОВАННЫЕ © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
15
СИНДРОМ МАРФАНА П Р О Я В Л Е Н И Я СИНДРОМ МАРФАНА П Р О Я В Л Е Н И Я * высокий рост * пролапс митрального клапана * вывих хрусталика ПОРАЖЕНИЕ СКЕЛЕТА ПОРАЖЕНИЕ СЕРДЦА И СОСУДОВ ПОРАЖЕНИЕ ГЛАЗ * дрожание радужки * аневризма аорты * длинные конечности * арахнодактилия * гиперподвижность суставов © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
16
Возможные наборы половых хромосом при нормальном и аномальном течении при нормальном и аномальном течении I мейотического деления I мейотического деления ХХХ Х ХХХY ХХХ полисомия Х ХО летальный ХХ летальный О ХХY норма норма? ХYХY О Y ХО ХYХY ОО ХYХYХХY YОYО с-м Клайнфельтера с-м Шерешевского- Тернера спермии яйцеклетки © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
17
Врождённый порок развития (ВПР) * Стойкое морфологическое изменение органа, его части или участка тела, * выходящее за пределы нормального диапазона строения. *Возникает внутриутробно. * Обусловливает расстройство жизнедеятельности организма. © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
18
ЭКТОПИЯ НАИБОЛЕЕ ЧАСТЫЕ ВРОЖДЕННЫЕ ПОРОКИ РАЗВИТИЯ НАИБОЛЕЕ ЧАСТЫЕ ВРОЖДЕННЫЕ ПОРОКИ РАЗВИТИЯ УДВОЕНИЕ/ УТРОЕНИЕ АПЛАЗИЯ, ГИПОПЛАЗИЯ ГЕТЕРОТОПИЯ СТЕНОЗ АТРЕЗИЯАГЕНЕЗИЯ ПЕРСИСТИРОВАНИЕ © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
19
ВИДЫ ВРОЖДЕННЫХ ПОРОКОВ РАЗВИТИЯ В ЗАВИСИМОСТИ ОТ ОБЪЕКТА ВОЗДЕЙСТВИЯ ПОВРЕЖДАЮЩИХ ФАКТОРОВ И СРОКА БЕРЕМЕННОСТИ ВИДЫ ВРОЖДЕННЫХ ПОРОКОВ РАЗВИТИЯ В ЗАВИСИМОСТИ ОТ ОБЪЕКТА ВОЗДЕЙСТВИЯ ПОВРЕЖДАЮЩИХ ФАКТОРОВ И СРОКА БЕРЕМЕННОСТИ ГАМЕТОПАТИИБЛАСТОПАТИИЭМБРИОПАТИИФЁТОПАТИИ *объект: половые клетки *объект: бластоциты *срок: первые 15 суток после оплодотворения *объект: эмбрион *срок: 16 день – 8-9 неделя беременности *объект: плод *срок: после 8-9 недели беременности © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
20
СВОЙСТВА МУЛЬТИФАКТОРИАЛЬНЫХ ЗАБОЛЕВАНИЙ П р и м е р ы: * Ишемическая болезнь сердца (ИБС) * Гипертоническая болезнь * Бронхиальная астма * Сахарный диабет * Язвенная болезнь желудка и кишечника * Псориаз * Эпилепсия * Системная красная волчанка *… в этиологии важна роль изменений в геноме предрасположенность реализуется под влиянием большого числа факторов среды характер наследования не объясняется только менделевскими законами предрасположенность к болезни зависит от большого числа генов (феномен аддитивности) © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
21
биологического моделирования генетики соматических клеток ОСНОВНЫЕ МЕТОДЫ ДИАГНОСТИКИ И АНАЛИЗА ПАТОГЕНЕЗА НАСЛЕДСТВЕННЫХ ФОРМ ПАТОЛОГИИ клинико- синдромологический клинико- генеалогический цито- генетический молекулярно- генетический близнецовый био- химический селекции блотгибридизации ДНК клонирования гибридизации гибридизация ДНК полимеразная цепная реакция клонирование ДНК © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
22
ПРИНЦИПЫ И МЕТОДЫ ЛЕЧЕНИЯ НАСЛЕДСТВЕННЫХ ФОРМ ПАТОЛОГИИ ПРИНЦИПЫ И МЕТОДЫ ЛЕЧЕНИЯ НАСЛЕДСТВЕННЫХ ФОРМ ПАТОЛОГИИ ЭТИОТРОПНЫЙПАТОГЕНЕТИЧЕСКИЙСИМПТОМАТИЧЕСКИЙ ПРИНЦИПЫ МЕТОДЫ * коррекция дефекта генома: – введение в геном нормального гена, – подавление репликации патогенного гена * изменение генома: – введение в него гена, кодирующего синтез чужеродного для системы ИБН антигена * заместительная терапия (введение дефицитного вещества) * коррекция метаболизма: – ограничения попадания в организм неметаболизируемых веществ (лактозы, фенилаланина), – выведение избытка метаболитов (холестерина, фенилпировиноградной кислоты), – регуляция активности ферментов (липопротеинлипазы крови, КФКазы) * устранение тягостных, усугубляющих состояние симптомов * хирургическое устранение дефектов (шунтов, сращений, создание шунтов) © П.Ф.Литвицкий, 2004 © ГЭОТАР-МЕД, 2004
Источник