
Синдром айкарди гутьереса тип 6

Что такое синдром Айкарди?
Синдром Айкарди — крайне редкое генетическое заболевание. Почти все люди с синдромом Айкарди — новорожденные девочки. У людей с синдромом Айкарди наблюдается агенезия мозолистого тела (отсутствие соединения между двумя полушариями мозга), хориоретинальные лакуны (дефекты в светочувствительной ткани в задней части глаза) и эпилептический припадок.
Синдром является спорадическим (случайным), что означает, что оно не передается от родителя к ребенку.
Причины
Синдром Айкарди, вероятно, возникает в результате новой мутации в гене, расположенном на Х-хромосоме. Точный ген или генетический механизм, вызывающий синдром Айкарди, пока неизвестен. Недавно обнаруженный отчет, описывающий изменения в генах TEAD1 и OCEL1 у двух девочек с синдромом, не был подтвержден в большой группе других девочек с синдромом Айкарди. Таким образом, эти гены, по-видимому, не являются причиной синдрома Айкарди. Предполагается, что это состояние является смертельным для новорожденных детей мужского пола.
Родители женщины с синдромом Айкарди обычно не страдают. О передаче синдрома Айкарди от пострадавшей матери ее ребенку не сообщалось. Другие члены семьи также обычно не подвергаются повышенному риску.
Симптомы и признаки
Синдром Айкарди обычно начинается с непроизвольных мышечных спазмов в возрасте от четырех месяцев до четырех лет.
Другие симптомы могут включать эпилепсию, умственную отсталость, глубокую мышечную слабость (гипотонию), аномально маленькую голову (микроцефалию), аномально маленькие глаза (микрофтальмию), неполное развитие сетчатки и нерва в задней части глаза (колобомы) и/или аномалии ребер и/или позвоночника.
У детей всех возрастов с синдромом Айкарди наблюдается значительная задержка моторного развития. Синдром Айкарди может быть опасным для жизни в детстве из-за осложнений инфекций верхних дыхательных путей.
Большая часть девочек с синдромом Айкарди имеет резкую задержку психомоторного развития. В неврологическом статусе часто отмечается — значительное уменьшение размеров черепа (микроцефалия), мышечная гипотония, возможна односторонняя мышечная гипертония и спастичность, оживленные глубокие сухожильные рефлексы или нарушение работоспособности конечностей (геми- или тетрапарез).
Агенезия мозолистого тела при синдроме Айкарди обычно тотальная, часто сопровождается с гетеротопией коркового вещества мозга, атрофией коры, структурной асимметрией полушарий мозга, нормотензивной гидроцефалией, полимикрогирией или пахигирией, хориоидальными кистами и папилломами, вентрикуломегалией, внутримозговыми кистами, синдромом Денди-Уокера.
Затронутые группы населения
Известно приблизительно о 500 случаях синдрома Айкарди во всем мире, особенно большое количество в Японии. Синдром встречается у детей с различной расовой принадлежностью.
По недавно проведенным в Швеции исследованиям распространенность синдрома Айкарди составляет от 2 до 15 случаев на 100000 девочек. В России аналогичные исследования к сожалению не проводились. Однако, учитывая фенотипическое разнообразие и диагностические трудности, многие случаи заболевания остаются недиагностированными. Это позволяет пересмотреть данные об истинной распространенности синдрома Айкарди в сторону увеличения, возможно, синдром Айкарди является более частой причиной задержки умственного развития и инфантильных спазмов у девочек, чем считается в настоящее время.
На сегодняшний день считается, что заболеваемость синдромом Айкарди среди всех детей с инфантильными спазмами составляет всего около 2-4%.
Диагностика
До настоящего времени не существует специального лабораторного диагностического теста или исследования, которое бы позволило поставить диагноз синдрома Айкарди. Для этого необходимо:
- неврологический осмотр;
- офтальмоскопия;
- электроэнцефалография (ЭЭГ);
- магнитно-резонансная томография с контрастом и/или без;
- рентгенограмма скелета.
На магнитно-резонансной томографии можно обнаружить агенезию мозолистого тела, асимметрию полушарий коры, гетеротопию коркового вещества, внутримозговые кисты, папиллому сосудистых сплетений и тд.. Аксоны коры, которые в норме должны перекрещиваться, при его агенезии не формируются и соответственно не идентифицируются при нейровизуализации.
Агенезия мозолистого тела позволяет боковым желудочкам распространиться вверх, во фронтальное и париетальное белое вещество. Это состояние именуется верхней транслокацией боковых желудочков в лобно-теменные регионы мозга. Аналогичное смещение вверх претерпевает и третий желудочек, что является одним из нейрорадиологических маркеров агенезии мозолистого тела. Увеличенный третий желудочек, выдвигаясь вперед и вверх, раздвигает передние рога боковых желудочков, при сопутствующей гидроцефалии объем желудочков увеличивается, задние рога расширяются и изгибаются по направлению к средней линии (форма «ухвата»). Вероятно отсутствие поддерживающей функции мозолистого тела является основой для типичной черты агенезии мозолистого тела — расширения полушарий, третьего желудочка и Монроева отверстия.
Типичные изменения на электроэнцефалографии в виде гипсаритмии, характерные для инфантильных спазмов встречаются далеко не у всех больных. Наиболее характерные изменения приступной ЭЭГ заключаются во вспышках нерегулярных быстрых и медленных волн продолжительностью от 3 до 6 секунд, которые перемежаются некоторым уплощением основного ритма в течение 5-20 секунд, причем изменения не синхронизированы по полушариям. На электроэнцефалографии — феномен «расщепленного мозга». Так как практически у половины (42%) пациентов инфантильные спазмы сочетаются с другими видами эпилептических приступов, то данные ЭЭГ могут быть противоречивы.
При офтальмоскопии обнаруживаются белого, или желто-белого цвета, хорошо отграниченные, круглые депигментированные участки.
Лечение синдрома Айкарди
Лечение синдрома Айкарди пока не разработано. В основном применяется симптоматическое лечение. Основная стратегия терапии — купирование инфантильных спазмов, которые зачастую резистентны к антиэпилептическим препаратам, лечение их сложное и эффективность его невелика. Применяют различные медикаменты в максимально высоких дозах.
Стартовая терапия начинается с вигабатрина (сабрил) — 50-100мг/кг/сут и вальпроатов (депакин-сироп) — 50-100мг/кг/сут. При частых приступах назначают комбинации антиэпилептических препаратов с бензодиазепинами (клоназепам) -0,25-2мг/сут, или фенобарбиталом(5-15мг/кг/сут), а так же может быть введен суксилеп 15-30мг/кг/сут.
Альтернативным методом является применение кортикостероидных гормонов (АКТГГ, синактен-депо в/м, дексаметазом, преднизолон) и октагам. Средняя дозировка преднизолона 1-2,5мг/кг/сут с последующим переходом на минимальную поддерживающую дозу. Гормоны назначают в сочетании с базовыми антиэпилептическими препаратами.
В качестве паллиативного хирургического лечения возможно использование стимуляции блуждающего нерва. Так как костно-мышечные дефекты могут приводить к сколиозу, для его предотвращения используется физиотерапия, лечебная физкультура, возможна хирургическая коррекция.
Прогноз
Прогноз для девочек с синдромом Айкарди варьируется в зависимости от тяжести их симптомов.
Прогноз при синдроме Айкарди, как правило, серьезен в связи с выраженной умственной отсталостью и резистентным характером судорог. Часть детей (до 25%) погибает в первые годы жизни. Из выживших детей только 25% самостоятельно ходят и только 50% имеют навыки самообслуживания.
Продолжительность жизни очень вариабельна, в зависимости от степени выраженности симптомов. Средняя продолжительность жизни по разным данным составляет от 8,3 до 18,5 лет. Но имеются сведения о женщине 32 лет с синдромом Айкарди, а так же 49 лет с умеренной формой синдрома
Источник
Синдром Айкарди-Гутьереса был впервые обнаружен Жаном Айкарди и Франсуазой Гутьер в 1984 году у 8 детей. Эти дети выявили аномалии или дефекты мозга, такие как аномалия в белом веществе и кальцификация мозговых ганглиев.
Мозговые ганглии расположены у подножия мозга, связаны с таламусом и стволом мозга.
Термин Синдром Айкарди-Гутьереса был введен в 1992 году, является чрезвычайно редким заболеванием. В 2001 году число выявленных случаев составило 50. Это число увеличилось с обнаружением мутаций в генах, связанных с этим синдромом. С этим синдромом связаны 6 основных генов.
Существуют другие гены, которые были идентифицированы с более низкой частотой. Симптомы во многом варьируются.
Наследование
Синдром Айкарди-Гутьереса является наследственным (аутосомно-рецессивным) состоянием, что означает, что болезнь передается генетически. Автосомальное рецессивное состояние возникает, когда обе копии гена или кластера генов являются дефектными.
Если плод наследует дефектные копии от обоих родителей, то возникает заболевание. В качестве альтернативы, плод может наследовать один дефектный ген от одного родителя, а оставшаяся нормальная копия повреждается из-за экологических причин, таких как вирусная инфекция или воздействие повреждающих ДНК агентов.

Рецессивное состояние указывает на то, что заболевание возникает только в случае дефектов обеих копий гена.
При доминирующей форме наследования только одна дефектная копия может вызвать проблему.
Автосомальное заболевание указывает, что состояние передается от одного поколения к другому через одну из 23 хромосом (аутосомы), а не половых хромосом (X или Y).
Синдром Айкарди наблюдается у младенцев в течение 4 месяцев после рождения и приводит к физической, умственной отсталости. Влияет на мозг, кожу, иммунную систему организма. Иммунный ответ подобен реакции организма на вирусную инфекцию, известен как «имитация врожденной инфекции».
При рождении типичными характеристиками являются неврологические аномалии, повышенный уровень белков печени в крови, увеличение печени, селезенки (гепатоспленомегалия), снижение уровня тромбоцитов крови (тромбоцитопения).
Существует два типа синдрома Айкарди, основанных на стадии возникновения. Ранняя стадия заболевания наблюдается у новорожденных, младенцев. Поздняя стадия заболевания наблюдается у детей старшего возраста (от 1 до 2 лет) с меньшим повреждением головного мозга.
Причины
Как упоминалось ранее, Синдром Айкарди является генетическим состоянием, которое наследуется аутосомно-рецессивным способом. В очень редких случаях синдром является преимущественно унаследованным. Мутация может возникать внезапно или спорадически.

Точный ген, который его вызывает, не совсем понятен. Гены, которые показывают мутации при этом расстройстве: SAMHD1, RNaseH2A, RNaseH2B, RNaseH2C, TREX1.
Ген SAMHD1 генерирует белок, который участвует при иммунном ответе. Дефекты приводят к иммунному отказу.
TREX1 расположен на хромосоме 3, называется AGS1. TREX1 кодирует белок DNase III, который режет одноцепочечную ДНК. RNaseH2A, RNaseH2B, RNaseH2C, кодируют ферменты, которые образуют белковый комплекс под названием RNAse2H, разрушают РНК. TREX1, RNaseH2A, RNaseH2B, RNaseH2C представляют собой гены, которые кодируют ферменты, называемые нуклеазами.
Они участвуют в разрушении ДНК и РНК. Когда есть дефекты в нуклеазах, существуют избыточные молекулы ДНК и РНК, заставляющие организм думать, что в нем присутствуют вирусные частицы. Это вызывает иммунную реакцию (защиту организма) против себя.
Последствиями реакции являются поражения, наблюдаемые на коже, дефекты головного мозга (энцефалопатия). Мутации ADAR6 также являются причиной расстройства.
Мутации RNaseH2C (AGS3) на хромосоме 11 наблюдаются в 40% случаев синдрома Айкарди-Гутьера. RNaseH2B (AGS2) на хромосоме 13 наблюдаются в 14%, RNaseH2A на хромосоме 19 (AGS4) – 4% случаев. Мутации TREX1 – 25% случаев.
TREX1, RNaseH2A, RNaseH2B способствуют развитию раннего синдрома Айкарди. RNaseH2B способствует позднему.
Существуют другие генетические нарушения, которые возникают из-за мутаций. Генетические расстройства, связанные с мутациями приведены ниже.

Генетическое расстройство, вызванное мутацией ADAR:
- Dyschromatosis symbolrica hereditaria 1 (DSH).
Генетические расстройства, вызванные TREX1:
- Семейная желчная волчанка;
- Критический энцефалит;
- Системная красная волчанка (СКВ);
- Аутосомно-доминантная вагиналопатия сетчатки с церебральной лейкодистрофией (RVCL).
Симптомы
При раннем начале синдрома Айкарди-Гутьерса симптомы появляются примерно через 4 месяца после рождения. Беременность нормальная, ребенок не проявляет никаких симптомов при рождении. Начальные признаки на этом этапе представлены ниже:
- Трудность кормления;
- Сильная раздражительность;
- Частые лихорадки без причины;
- Ошибочные режимы сна;
- Приступы или эпилепсия.
Существуют дефекты развития мозга, что приводит к замедлению роста головы, ухудшению тонких моторных навыков, вообще замедлению неврологического развития.
Эти симптомы описаны ниже:
- Реакция испуга даже на мягкие раздражители;
- Только частичное управление головкой;
- Аномалии движения глаз и детских рефлексов (например, сосание);
- Языковое общение также ухудшается.
Эти симптомы сохраняются в течение нескольких месяцев, затем стабилизируются или исчезают. Существует прекращение прогрессирования заболевания.
В фазе позднего начала Синдрома Айкарди-Гутье, эти симптомы появляются только через 1 или 2 года после нормального физического развития и развития мозга.
20% случаев признаки появляются у новорожденных.
В матке могут быть обнаружены типичные особенности увеличенной печени и селезенки, повышенные уровни ферментов печени в крови, аномальное образование головного мозга, снижение тромбоцитов крови (тромбоцитопения), анемия.
Другие пострадавшие органы – кожа, печень, глаза, дефициты гормонов и иммунной системы.
- Поражения кожи, воспаление, рубцевание ткани (например, уши, пальцы ног, пальцы);
- Глаукома;
- Инсулинзависимый сахарный диабет;
- Дефицит антидиуретического гормона (гормональное мочеиспускание);
- малокровие;
- Антитела против иммунной системы (аутоантитела);
- Малая голова (микроцефалия);
- Непроизвольные мышечные сокращения, влияющие на осанку, движение.
Диагностика
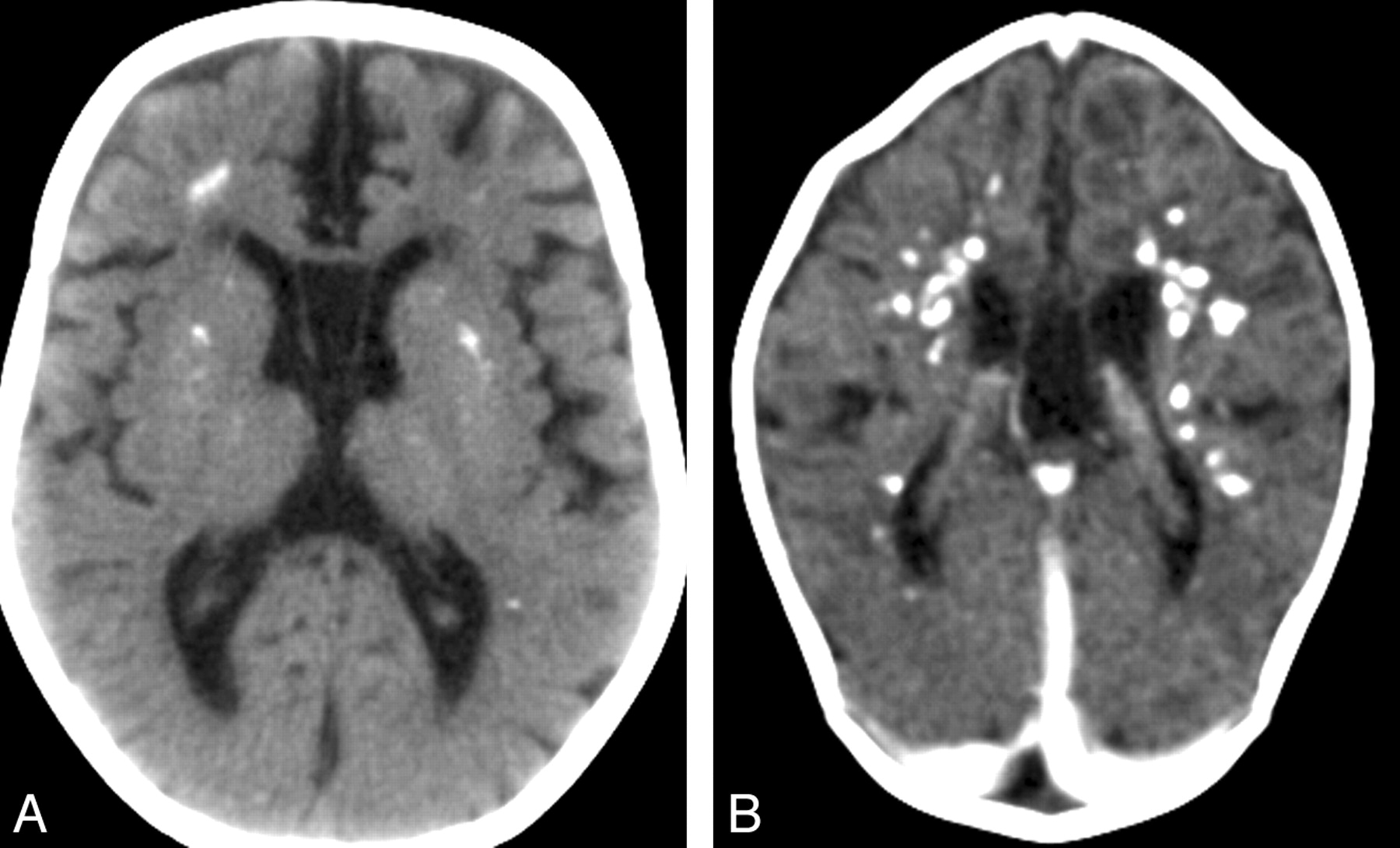
Синдром Айкарди-Гутьера можно диагностировать на основании клинических симптомов с использованием компьютерной томографии (КТ), магнитно-резонансной томографии (МРТ), анализа генов.
Существуют определенные характерные особенности, которые могут быть использованы для конкретной диагностики людей с синдромом Айкарди-Гутье. Они заключаются в следующем:
- Увеличение концентрации неоптерина в спинномозговой жидкости (CSF);
- Больше кальцификации ганглиев в головном мозге;
- Увеличение концентрации интерферона-альфа (IFN-a);
- Церебральная атрофия;
- Интерферонная подпись;
- Изменения в белом веществе;
- Увеличение количества белых кровяных телец (лейкоцитоз).
КТ-сканирование помогает визуализировать кальцификацию головного мозга. Атрофия мозга, изменения в белом веществе, лейкоцитоз, другие изменения обнаруживаются с помощью МРТ.
Количественная ПЦР используется для обнаружения интерфероновой сигнатуры. Это очень надежный тест, поскольку может обнаруживать экспрессию интерфероно-стимулированных генов крови.

Гены видны даже после того, как симптомы синдрома Айкарди-Гутьера сократились.
Неоптериновые концентрации в CSF являются надежным источником для подтверждения диагноза.
Индивидуальное тестирование для 6 основных генов, которые вызывают синдром Айкарди-Гутье, полезно для подтверждения диагноза.
Тестирование мультигенных панелей является более эффективным и надежным для подтверждения диагноза. Мультигенная панель анализирует все 6 генов одновременно.
Пренатальное тестирование включает взятие пробы крови (PUBS), предимплантационную генетическую диагностику, генетическое тестирование. Кровь тестируется у плода в третьем триместре.
При предимплантационной генетической диагностике семьи, у которых есть подозрение о наличии причинных генов синдрома Айкарди, используют генный тест для эмбриона.
Лечение
Существует несколько вариантов лечения, так как это относительно новый синдром. Исследования все еще распутывают его детали. Симптомы лечатся, однако нет лекарств от заболевания. Основываясь на текущих знаниях, были разработаны следующие методы лечения:
Обработка ингибитора обратной транскриптазы (RTI)
Пациенты с синдромом Айкарди-Гутьере могут воспользоваться обработкой RTI, поскольку TREX1 и SAMHDI являются нуклеазами, которые переваривают ДНК. В отсутствие этих генов происходит накопление ДНК, которая замедляется ингибиторами обратной транскриптазы. Эти ИРТ используются для лечения пациентов с ВИЧ, их пределы безопасности четко известны.
Антитела против интерферона альфа
Анти-интерферон-альфа-антитела используются для блокирования активности рецептора интерферона альфа-типа I или различных форм интерферона-альфа в CSF.
Препараты против аутоантител
Синдром Айкарди-Гутьера приводит к иммунному ответу, когда продуцирование антител увеличивается против собственных клеток организма. Лекарства, направленные против увеличенных В, Т-клеток, являются формой лечения.

Миофенолатный мофетил используется против реакционноспособных Т-клеток.
Такие лекарства имеют побочные эффекты, однако их использование контролируется на основе степени прогрессирования заболевания.
Другие формы лечения
- Лечение фолиновой кислотой увеличивает уровень фолата в CSF.
- При активном иммунном ответе, вызванном синдромом Айкарди-Гутье, рекомендуется терапия кортикостероидами.
Советы по здоровью
Приступы могут контролироваться стандартными процедурами и лекарствами.
Адекватное внимание следует уделять методу питания, типу потребляемой пищи. Необходимо поддерживать адекватное потребление калорий для надлежащего развития.
Подходящее лечение патологий грудной клетки и физиотерапия – еще один способ справиться с осложнениями, которые возникают из-за легочных инфекций при синдроме Айкарди-Гутье.
Генетическая консультация рекомендуется в семьях с высоким риском, чтобы выяснить наличие носителей мутаций генов. Младенцы должны регулярно контролироваться для риска появления глаукомы, сахарного диабета, неправильного развития позвоночника.
К какому врачу обратиться
Когда ребенок показывает типичные симптомы припадков, раздражительность, неустойчивые сон, следует проконсультироваться с педиатром. Основываясь на симптомах и диагнозе, ребенок будет передан неврологу.
Различные термины
Кри энцефалит; AGS; семейная инфантильная энцефалопатия с хроническим лимфоцитозом цереброспинальной жидкости и внутричерепной кальцификацией; синдром псевдотоксикоплазмоза; энцефалопатия с классификацией базальных ганглиев; синдром псевдо-TORCH.
Продолжительность жизни
Около 25% лиц с синдромом Айкарди-Гутьера умирают до 17 лет.
Понравилась статья? Поделись с друзьями:
Источник
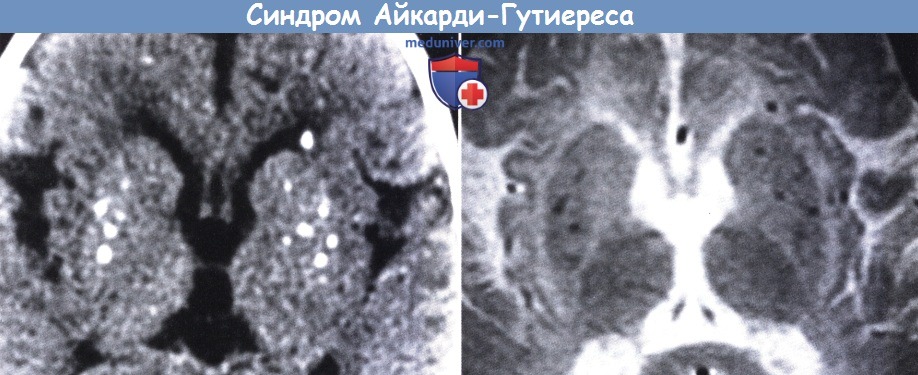
Лейкодистрофия с кальцификацией базальных ганглиев и лимфоцитозом спинномозговой жидкости (синдром Айкарди-Гутиереса)Лейкодистрофия с кальцификацией базальных ганглиев и лимфоцитозом спинномозговой жидкости (синдром Айкарди-Гутиереса) заболевание является аутосомно-рецессивным, патологическая анатомия изучена плохо, но свидетельствует о микроваскулите. В сходных случаях помимо воспалительной реакции менингеальных оболочек обнаруживалась дополнительная очаговая лейкодистрофия с сохраненными островками миелина. Клинические проявления включают очень раннее начало в первые недели или месяцы жизни, таким образом, явная деградация отсутствует. Общее состояние пациентов плохое, отмечается заметная гипотония, чередующаяся с эпизодами опистотонуса, нарушением развития, фебрильными эпизодами; смерть в состоянии децеребрации наступает в течение нескольких лет, однако некоторые дети живут дольше (Aicardi и Goutieres, 1984; Goutieres et al, 1998). Вместе с тем, описаны случаи позднего начала в возрасте 6-12 месяцев, когда возможно менее тяжелое течение заболевания (Rice et al, 2007). Два основных диагностических проявления: 1) наличие кальцификатов в базальных ядрах, а иногда и в перивенрикулярном белом веществе и в зубчатом ядре в сочетании с пониженной плотностью белого вещества и атрофией головного мозга; 2) устойчивый умеренный лимфоцитоз спинномозговой жидкости (10-80 клеток/мм3), который носит непостоянный характер и имеет тенденцию со временем снижаться. Повреждения кожи (подобные обморожениям) отмечаются примерно в половине случаев. В большинстве случаев отмечается умеренное повышение интерферона-альфа в спинномозговой жидкости, как минимум в первый год заболевания (Lebon et al., 1988), данное проявление менее выражено в крови.
Сходные случаи были зарегистрированы как «энцефалит Кри», встречающийся в высоко инбредных в Северном Квебеке с высокой частотой близкородственных браков (Black et al., 1988). Синдром Айкарди-Гутиерреса является генетически гетерогенным заболеванием, выявлено четыре различных локуса, связанных с развитием симптомов. Недавно были продемонстрированы мутации субъединиц четырех генов, кодирующих рибонуклеазу TREX1 (или хромосомы 3q21), и генов, кодирующих три субъединицы рибонуклеазы белкового комплекса H2 (Crow et al., 2006; Rice et al., 2007). Следует отличать заболевание от врожденных вирусных инфекций, в особенности вызванных цитомегаловирусом и вирусом иммунодефицита человека от случаев кальцификации базальных ганглиев без признаков поражения белого вещества, иногда в сочетании с атрофией головного мозга и без плейоцитоза, который может изначально отсутствовать или исчезает в дальнейшем. Такие случаи нередки (Billard et al., 1989) и, вероятно, представляют собой последствия нескольких непрогрессирующих негенетических заболеваний, которые, тем не менее, также могут относиться к метаболическим болезням. В таких случаях всегда следует осуществлять поиск герпес-вирусной инфекции. – Также рекомендуем “Синдром Коккейна – причины, диагностика, лечение” Редактор: Искандер Милевски. Дата публикации: 17.12.2018 Оглавление темы “Дегенеративные заболевания нервной системы.”:
|
Источник