
Синдром беквита видемана фото взрослых

Синдром Беквитта-Видемана: причины, диагностика, лечениеЭтиология и встречаемость синдрома Беквитта-Видемана. Синдром Беквитта-Видемана (MIM №130650) — панэтнический синдром, обычно спорадический, но иногда может наследоваться как аутосомно-доминантный. Синдром Беквитта-Видемана встречается приблизительно у одного из 13 700 живорожденных. Синдром Беквитта-Видемана вызван нарушением баланса экспрессии импринтированных генов в регионе р15 хромосомы 11. Эти гены включают транскрибируемые, но не транслируемые KCNQOT1 и Н19, и кодирующие белки гены CDKN1C и IGF2. В норме эти гены импринтированы и экспрессируются только из отцовского (IGF2 и KCNQOT1) или только материнского аллеля (HI9 и CDKN1C). IGF2 кодирует инсулиноподобный фактор роста, стимулирующий рост; CDKN1C кодирует супрессор клеточного цикла, ограничивающий деление и рост клеток. Транскрипция РНК Н19 и KCNQOT1 подавляет экспрессию материнской копии IGF2 и отцовской копии CDKN1С соответственно. Несбалансированная экспрессия импринтированных генов в 11р15 может происходить по множеству механизмов. Мутации в материнском аллеле CDKN1C обнаруживают в 5-10% спорадических случаев и в 40% семей с аутосомно-доминантным синдромом Беквитта-Видемана. Большинство пациентов с синдром Беквитта-Видемана, тем не менее, имеют снижение экспрессии материнского аллеля гена CDKN1С вследствие аномального импринтинга, а не мутации. У 10-20% индивидуумов с синдромом Беквитта-Видемана снижение экспрессии материнской копии CDKN1C и повышение экспрессии IGF2 вызвано отцовской изодисомией 11р15. Поскольку соматическая рекомбинация, ведущая к сегментной однородительской дисомии, происходит после зачатия, больные с сегментной однородительской дисомией — мозаики, и для выявления изодисомии может потребоваться исследование других тканей, кроме крови. Еще 1-2% больных с синдром Беквитта-Видемана имеют цитогенетически обнаруживаемую хромосомную аномалию, например, материнскую транслокацию, инверсию хромосомы 11 или дупликацию отцовской хромосомы 11р15. Таким образом, чтобы исключить структурную аномалию 11р15 при проведении генетического консультирования, необходимо кариотипирование родителей. При синдроме Беквитта-Видемана также обнаружены редкие микроделеции в гене KCNQOT1 или Н19, нарушающие импринтинг. У остальных пациентов аномалии в импринтинге и экспрессии генов остаются невыясненными.
Патогенез синдрома Беквитта-ВидеманаВ ходе гаметогенеза и в раннем зародышевом развитии у мужчин и женщин устанавливаются различные типы метилирования ДНК в генах KCNQOT1 и Н19. Аномальный импринтинг при синдроме Беквитта-Видемана легче всего обнаружить при анализе метилирования ДНК в специфических участках CpG в генах KCNQOT1 и Н19. У 60% лиц с синдромом Беквитта-Видемана обнаруживают гипометилирование материнского аллеля KCNQOT1. У других 2-7% больных гиперметилирование материнского гена Н19 снижает его экспрессию, что приводит к избыточной экспрессии IGF2. Несоответствующая экспрессия обоих родительских аллелей IGF2 может объяснить избыточный рост, наблюдаемый при синдроме Беквитта-Видемана. Аналогично снижение экспрессии материнской копии CDKN1С удаляет ограничение роста плода. Фенотип и развитие синдрома Беквитта-ВидеманаСиндром Беквитта-Видемана связан с пренатальным и постнатальным избыточным ростом. До 50% больных рождаются недоношенными и превышают массу тела, соответствующую гестационному сроку при рождении. Плацента также увеличена, а беременность часто осложняется многоводием. Кроме этого, у новорожденных с синдром Беквитта-Видемана часто бывают такие осложнения, как омфалоцеле, макроглоссия, неонатальная гипогликемия и кардиомиопатия, приводящие к 20% смертности. Неонатальная гипогликемия обычно мягкая и непостоянная, но есть сообщения о некоторых случаях более серьезной гипогликемии. Пороки развития почек и повышение кальция в моче с развитием нефрокальциноза и мочевых камней отмечают почти у половины больных. Гиперплазия различных сегментов тела или отдельных органов может выявляться уже при рождении и становится более или менее заметной со временем. Развитие больных обычно нормальное, если у них нет несбалансированной хромосомной аномалии. Дети с синдромом Беквитта-Видемана имеют повышенный риск развития эмбриональных опухолей, особенно опухоли Вильмса и гепатобластомы. Общий риск новообразований у детей с синдромом Беквитта-Видемана приблизительно 7,5%; риск значительно снижается после достижения детьми 8 лет. Особенности фенотипических проявлений синдрома Беквитта-Видемана: Лечение синдрома Беквитта-ВидеманаОказание помощи детям с синдромом Беквитта-Видемана включает лечение имеющихся симптомов, например коррекцию омфалоцеле и гипогликемии. Макроглоссия может потребовать специальных методов вскармливания или занятий с логопедом. При крупных дефектах брюшной стенки, асимметрии длины ног и при пороках развития почек может оказаться необходимым хирургическое вмешательство. Если имеется гиперкальциурия, может быть назначена терапия, направленная на уменьшение выделения кальция. Важно периодическое обследование на эмбриональные опухоли, поскольку они отличаются быстрым ростом и злокачественностью. Текущие рекомендации для исключения опухолей — УЗИ брюшной полости каждые 3 мес в течение первых 8 лет жизни и измерение сывороточного АФП каждые 6 нед в течение первых нескольких лет жизни.
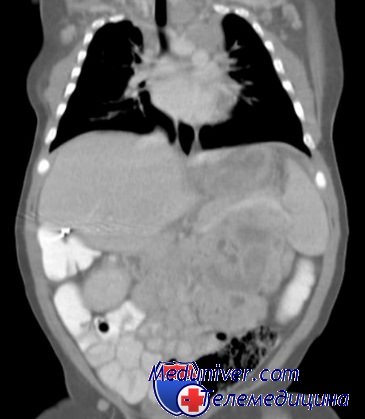
Риск повторения – наследования синдрома Беквитта-ВидеманаРиск повторения для сибсов и потомства детей с синдромом Беквитта-Видемана существенно изменяется в зависимости от молекулярной основы заболевания. См. таблицу риска повторения для различных молекулярных изменений. Повышение риска синдрома Беквитта-Видемана при применении вспомогательных репродуктивных технологийВспомогательные репродуктивные технологии, например ЭКО и ИКСИ, становятся обычной процедурой, составляющей теперь во многих странах до 1-2% всех рождений. Ретроспективные исследования показали, что при беременностях, закончившихся новорожденными с синдромом Беквитта-Видемана, ЭКО использовалось в 10-20 раз чаще по сравнению с контролем. Риск синдрома Беквитта-Видемана после ЭКО оценивают как 1 на 4000, что в 9 раз выше, чем в общей популяции. Причина повышенной встречаемости дефектов импринтинга после ЭКО неизвестна. Встречаемость синдрома Прадера-Вилли, дефекта отцовского импринтинга, после ЭКО не повышена, а частота синдрома Ангельмана, дефекта материнского импринтинга, после ЭКО повышается, что позволяет предположить специфические отношения между ЭКО и материнским импринтингом. Поскольку отцовский импринтинг происходит задолго до ЭКО, а материнский происходит значительно ближе ко времени оплодотворения, роль ЭКО, как предрасполагающего к дефектам импринтинга фактора, требует серьезного анализа. Пример синдрома Беквитта-Видемана. А.Б., 27-летняя беременная, обратилась в пренатальный диагностический центр для проведения ультрасонографии 2-го уровня и генетического консультирования после планового УЗИ, обнаружившего крупный для данного гестационного возраста мужской плод с возможным омфалоцеле. Беременность, первая у каждого из родителей, наступила самопроизвольно, без вспомогательных репродуктивных технологий. После обследования ультрасонографией 2-го уровня семье выдано заключение, что плод имеет множество аномалий, наиболее соответствующих диагнозу синдрома Беквитта-Видемана, хотя не исключены и другие врожденные дефекты. Семейная пара решила не подвергаться процедуре амниоцентеза. Младенец родился кесаревым сечением в 37 нед с массой тела при рождении 9 фунтов и 2 унции (4 кг 140 г) и заметно увеличенной плацентой. Отмечены омфалоцеле, макроглоссия и вертикальные складки на мочках ушей. Консультант-генетик поставил клинический диагноз синдрома Беквитта-Видемана. После развития гипогликемии ребенок переведен в палату интенсивного наблюдения и в течение 1 нед получал внутривенные вливания глюкозы; гипогликемия разрешилась спонтанно. Результаты оценки сердечно-сосудистой деятельности нормальные, омфалоцеле откорректировано хирургическим путем без осложнений. Исследование метилирования гена KCNQOT1 подтвердило дефект импринтинга в 11р15, соответствующий диагнозу синдрома Беквитта-Видемана. Для исключения опухоли Вильмса рекомендовано каждые 3 мес УЗИ органов брюшной полости до достижения 8 лет и определение сывороточного АФП каждые 6 нед, как скрининговое обследование на гепатобластому, в течение первых 3 лет жизни. При последующих визитах, с учетом отрицательного семейного анамнеза и нормальных кариотипов родителей, дефект импринтинга в этой семье был расценен как спорадический случай синдрома Беквитта-Видемана с низким риском повторения. – Также рекомендуем “Наследственный рак молочной железы и яичников: причины, диагностика, лечение” Оглавление темы “Наследственные болезни”:
|

Источник
Пьебалдизм
Пьебалдизм похож на витилиго, поскольку оба заболевания характеризуются нарушением обмена пигментов и появлением белых прядей волос. Однако пьебалдизм отличается локализацией пятен на коже груди и живота. Кроме того, его признаки — наличие локона белых волос, более темных пятен на лишенной пигмента коже. Эти особенности видны уже с рождения.

Пьебалдизм является редкой наследственной болезнью, а частота ее распространения составляет всего 1 случай на 17 тысяч человек. Эта аномалия никак не влияет на работу внутренних органов и систем.
Анизокория
Анизокория — это состояние, при котором у человека наблюдается разница между размерами зрачков. При этом часто один зрачок имеет патологию, а именно, постоянно находится в зафиксированном положении.

Клинически важно установить, какой именно из зрачков находится в патологическом состоянии, так как это может указывать на наличие других отклонений. Например, отсутствие активности у меньшего из двух зрачков может быть признаком синдрома Горнера, который возникает при поражении нервной системы.
Гетерохромия
Еще одна аномалия, связанная с глазами — гетерохромия. Она выражается в различном цвете радужной оболочки правого и левого глаза или разной окраске определенных участков глаз. Возникает это нарушение от избытка или недостатка меланина — пигмента, который отвечает за цвет. У альбиносов он, к примеру, может вообще отсутствовать, а у людей с гетерохромией наблюдается в измененном состоянии.

Эта аномалия настолько популярна, что многие даже покупают разноцветные линзы, подражая людям с гетерохромией. А еще данная особенность очень привлекает представителей модельных агентств. Мы уже рассказывали о двух братьях из Турции, которые благодаря гетерохромии стали моделями.
Витилиго
О витилиго вы, наверное, уже наслышаны. Сейчас очень много моделей с этим заболеванием, и когда-то мы показывали вам фото самых популярных из них. Витилиго — это нарушение пигментации, которое выражается в исчезновении пигмента меланина на отдельных участках кожи.

Витилиго может начаться в любом возрасте в результате воздействия некоторых лекарственных и химических веществ, после воспалительных и некротических процессов на коже, из-за нервно-трофических, нейроэндокринных и аутоиммунных факторов. А вот предрасположенность к витилиго передается по наследству.
Синдром Шмида-Фраккаро
Синдром Шмида-Фраккаро, который также называют Синдромом кошачьего глаза — редкая врожденная патология, которая возникает из-за наличия дополнительной хромосомы. Один из характерных клинических признаков синдрома — верхняя коломба глаза, то есть отсутствие части радужной оболочки. Этот дефект делает взгляд человека похожим на кошачий, отсюда и название.
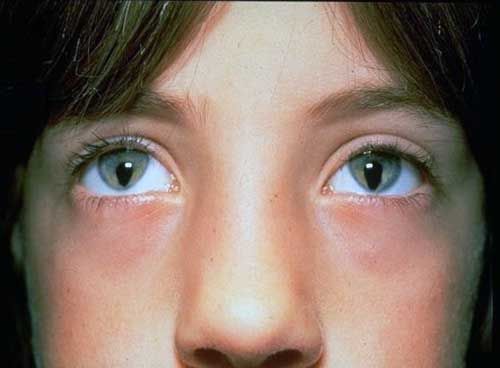
Особенность внешности — далеко не единственное, с чем приходится столкнуться больным синдромом Шмида-Фраккаро. У них могут наблюдаться пороки сердца, низкорослость, аномалии развития почек, умственная отсталость, сколиоз и многие другие проблемы.
Синдром нерасчесываемых волос
Синдром нерасчесываемых волос (еще одно его название — стекловидные волосы) делает его носителей похожими на одуванчики. Их шевелюра торчит во все стороны, при этом возможность уложить ее, собрать в пучок или заплести косу отсутствует в связи с огромным объемом, вызванным структурной аномалией.
Обычно заболевание развивается в грудном возрасте, а в период полового созревания проходит само. Но медицине известны случаи, когда этот синдром преследовал людей на протяжении всей жизни. И хоть сама по себе эта аномалия не представляет никакой опасности, согласитесь, приятного мало.
Смотрите также — «Дети не твои, и ты — не ты», или Как нерожденная женщина стала матерью четырех детей
Понравилось? Хотите быть в курсе обновлений? Подписывайтесь на наш Twitter, страницу в Facebook или канал в Telegram.
Источник
Источник
Синдром Беквита-Видемана представляет собой заболевание, характеризующееся широким спектром симптомов и физических признаков, которые варьируются по степени тяжести. Заболевание является врожденным и диагностировать можно еще на стадии беременности. Однако, в некоторых случаях наблюдается средний вес ребенка при рождении и скоростное увеличение роста после рождения, называется данная аномалия развития макросомией, необычайно большой язык — макроглоссия, увеличение некоторых внутренних органов органомегалия, и дефекты брюшной стенки омфалоцеле или пупочная грыжа.
Синдром Беквита Видемана может быть связан с низкий уровнем сахара в крови в течение первых нескольких дней (неонатальная гипогликемия), отличительными признаками мочек ушей и другие лицевые аномалии, аномальное увеличение одной стороны или строение тела в результате неравномерного (асимметричного) роста, и повышенным риском развития некоторых детских онкологических заболеваний, наиболее часто опухоли Вильмса и гепатобластома. Примерно 85 процентов людей с данным синдромом имеют генетические изменения, которые, как представляется, происходят случайным образом. Синдром также передаётся по наследству, но составляет не большой %, около 10-15% людей с этим синдромом.
Изначально данный синдром назывался по-другому и включал в себя несколько заболеваний: омфалоцеле, макроглоссия, синдром гигантизма. Но в 1969 году сочетание врожденных аномалий было переименовано в синдром Беквита-Видемана.

Признаки и симптомы
Симптомы синдрома Беквита-Видемана настолько различны, что назвать хотя бы пару объединяющих симптомов или признаков, которые встречаются у больше половины лиц с данным синдромом не получится. Диагностика синдрома Беквита-Видемана может быть осложнена, так как пациенты часто рождаются с генетическими изменениями, происходящим в отдельных клетках или участках тела.
Многие клинические особенности синдрома Беквита-Видемана в период развития ребенка могут стать менее заметными, а уже в более взрослом возрасте многие могут прийти к нормальному росту и внешнему виду.
Данный синдром как правило не затрагивает интеллектуальные способности ребенка, особенно если не связано с длительной гипогликемией или хромосомной дупликацией.
Некоторые младенцы с синдромом Беквита-Видемана рождаются раньше назначенного срока, и особенность в том, что недоношенные малыши уже имеют избыточный вес. Рост продолжается в детском возрасте и замедляется в возрасте от 7 до 8 лет. Патологическое расширение одной стороны или строение тела может привести к неравномерному росту.
Дефект передней брюшной стенки выглядит как пупочная грыжа (также известный как омфалоцеле), в котором часть кишечника ребенка и органы брюшной полости выпячиваются и торчат через пупок. Кишечник и другие органы покрыты тонкой мембраной. Менее серьезные дефекты могут выглядеть как выступание части кишечника через аномальное отверстие в мышечной стенке живота вблизи пуповинной грыжи. Внутренние органы младенцев могут быть аномально увеличенными, могут быть затронуты некоторые из перечисленных органов:
- Печень
- Селезенка
- Поджелудочная железа
- Почки или надпочечники
Некоторые новорожденные с синдромом Беквита-Видемана могут иметь низкий уровень сахара в крови из-за своей аномалии в развитии, а именно слишком большого роста и чрезмерной секреции гормона инсулина островками поджелудочной железы.
У детей с синдромом Беквита-Видемана часто наблюдается увеличенный в размерах язык (макроглоссия), из-за этого могут появляться трудности при разговоре, кормление или дыхании.
Помимо увеличенного языка синдром может иметь и другие аномалии черепа и лица (черепно-лицевой) области. К таким особенностям могут относиться отличительные складки в области ушей и вмятины на задних отделах ушей, и в задней область черепа.
Некоторые младенцы могут иметь бледно-красные или красновато-фиолетовые пятна на лица при рождении, чаще всего на веках и лбу, появляются данные пятна из-за большого скопления мелких кровеносных сосудов. Пятна обычно проходят или становятся менее заметными в течение первого года жизни.
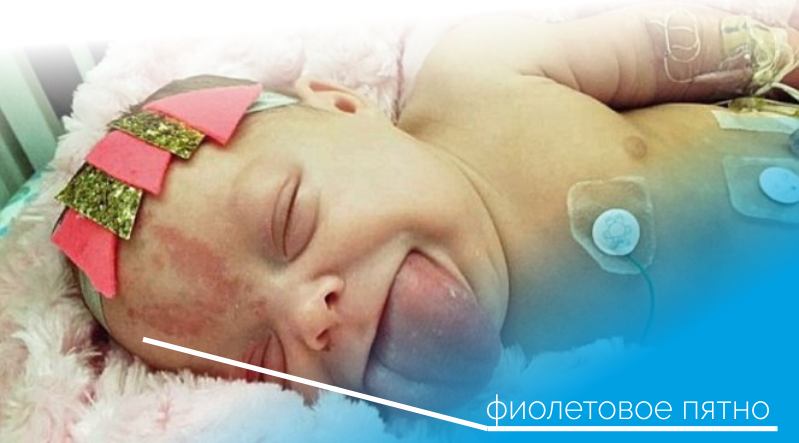
Также у детей можно заметить, что одна сторона лица больше, чем другая, а также гетерохромию (разный цвет глах).
Кроме того, у некоторых детей может наблюдаться неправильное соприкосновение верхних и нижних зубов или выступ нижней челюсти, особенно часто это наблюдается у детей с увеличенным языком.
Помимо перечисленных симптомов и признаков у детей с синдромом Беквита-Видемана, наблюдается неправильное развитие почек или почечная медуллярная дисплазия, и образование кальциевых отложений в почках (нефрокальциноз), которые могут ухудшить работу почек.
Дети с синдромом беквита-видемана могут иметь повышенный риск развития раковых заболеваний, чаще это нефробластома, заболевание представляет собой злокачественное новообразование почки, и опухоли с вовлечением печени (гепатобластома). Реже встречаются другие злокачественные новообразования такие как рабдомиосаркома. Появление злокачественных новообразований наиболее возможно до 8 лет.
Причины
Около 85 процентов детей, рожденных с синдромом Беквита-Видемана, не имеют наследственной истории заболевания, поэтому считать синдром наследственным заболеванием нельзя, но тот факт, что рождаются дети от родителей с тем же синдромом не дает возможности отрицать это.
Синдромом Беквита-Видемана результат различных патологий, влияющих на правильную экспрессию генов, которые контролируют рост в определенной области 11 хромосомою (11p15.5 — регуляция роста).
У каждого ребенка имеется по два экземпляра каждого гена, по одному от отца и по одному от матери. Правильный геномный отпечаток необходим для нормального развития и дефектный отпечаток на хромосоме 11 может привести к такому синдрому. Несколько генов, которые контролируют рост на одиннадцатой хромосоме отпечатываются, что означает, что активен только один ген от матери или хромосомы отца, а не обоих как должно быть.
Исследования показывают, что у детей, зачатых с помощью вспомогательных репродуктивных технологий, таких как экстракорпоральное оплодотворение (ЭКО) может быть более высокий риск развития расстройства в результате геномного импринтинга.
Диагностика заболевания
Синдром Беквита-Видемана может быть диагностирован и подтвержден после рождения на основе тщательного клинического обследования, выявления характерных физических признаков (например, увеличения веса или роста, макроглоссии)
В ряде случаев некоторые процедуры могут быть выполнены до рождения. Например, УЗИ может определить размеры развивающегося плода и показать другие признаки, что может свидетельствовать о синдроме Беквита-Видемана, например, увеличение амниотической жидкости, увеличенная плацента, омфалоцеле, увеличена окружность живота, или другие аномалии.
Лечение
Лечение синдрома Беквита-Видемана направлено на лечение конкретных признаков и симптомов, которые проявляются у каждого индивидуально. Лечение может потребоваться у разных специалистов одновременно, генетики, педиатры, пластические хирурги, специалисты по почкам, стоматологи, речевые патологи, педиатрические онкологи и другие специалисты в области здравоохранения.
У новорожденных с синдромом Беквита-Видемана, необходимо постоянно контролировать уровня сахара в крови, чтобы обеспечить своевременное выявление и лечение гипогликемии.
У многих младенцев с пупочной грыжей дефект может неожиданно исчезнуть к возрасту примерно один год. Операция, как правило, не требуется, если пупочная грыжа не увеличивается. Однако, у новорожденных с омфалоцеле, хирургическая коррекция дефекта, требуется сразу после рождения.
В дополнение к консультациям с пластическими хирургами и пульмонологами детям с макроглоссией следует пройти исследование сна, а также изучить процесс приема пищи. Трудности с питанием, вызванные аномально большим языком. Некоторым детям может потребоваться операция по уменьшению языка. Такая операция проводится, если макроглоссия вызывает дефекты строения десен и зубов, затруднения глотания, или мешает при разговоре. Имеются случаи, когда макроглоссия проходила сама, без хирургического вмешательства.
Кроме того, младенцы с синдромом Беквита-Видемана должны проходить регулярные обследования брюшной полости и почек, это рекомендовано для раннего выявления и лечения злокачественных опухолей, которые могут возникнуть в связи с данным синдромом.
Источник