
Синдром блума что это такое

Синдром Блума или Синдром Блум-Торре-Мачэйкик — редкое аутосомно-рецессивное заболевание, для которого характерны низкий рост больных и предрасположенность к онкологическим заболеваниям[2][3][4][5]. Клетки больных синдромом Блума демонстрируют сильную геномную нестабильность, в частности, частую гомологичную рекомбинацию. Заболевание было обнаружено и впервые описано американским дерматологом доктором Дэвидом Блумом в 1954 году[6].
Описание[править | править код]
Синдром Блума — разновидность прогероидных синдромов и демонстрирует ряд признаков, характерных для этой группы заболеваний: короткий рост и рано развивающаяся сыпь на областях кожи, контактирующих с солнечным светом. Кожная сыпь эритемная, склеродермическая и масштабная. При поражении кожи в области носа и щек, сыпь имеет бабочкообразную форму. Сыпь также может развиться на других поверхностях, контактирующих с солнечным светом, например, коже рук. Другие клинические проявления включают: высокий голос; характерные черты лица, такие как длинное, узкое лицо, микрогнатия, и выступающие нос и уши; гипо- и гиперпигментацию кожи; склеродермия, которая может проявляться как на коже, так и в глазах; умеренный иммунодефицит, связанный с недостатком определённых классов иммуноглобулинов, что приводит к увеличенному риску развития пневмонии и ушных инфекций; гипогонадизм.
Осложнения заболевания могут включать хронические проблемы с лёгкими, диабеты и неспособность к обучению. У небольшого количества больных отмечена умственная отсталость.
Самым существенным осложнением является увеличенный риск развития онкологических заболеваний.
Для синдрома Блума характерны некоторые фенотипы, общие для него с анемией Фанкони, что может быть следствием перекрытия функций мутантных белков, связанных с этими заболеваниями[7].
Связь с раком[править | править код]
Повышенный уровень мутаций при синдроме Блума приводит к высокому риску развития рака у пациентов[8]. Предрасположенность к раку характеризуется, во-первых, широким спектром типов рака, включая лейкозы, лимфомы и карциномы; во-вторых, ранним возрастом развития опухоли, по сравнению со средним возрастом в здоровой популяции; в-третьих, множественностью[9].
Белок синдрома Блума[править | править код]
Белок синдрома Блума — белок, кодируемый у людей геном BLM, и который не экспрессируется в случае наличия синдрома Блума[10].
Продукт гена BLM связан с подмножеством DExH box-содержащих хеликаз и имеет как ДНК-стимулированную АТФазную активность, так и АТФ-зависимую ДНК хеликазную активность. Мутации, вызывающие синдром Блума, изменяют или приводят к удалению хеликазного мотива и могут полностью убрать 3′ → 5′ хеликазную активность. В норме этот белок может выступать супрессором неверной гомологичной рекомбинации[11].
Взаимодействия[править | править код]
Для белка синдрома Блума была продемонстрирована возможность взаимодействия с CHEK1,[12]Replication protein A1,[13][14][15]Werner syndrome ATP-dependent helicase,[16]RAD51L3,[17]Ataxia telangiectasia mutated,[18][19]RAD51,[20]XRCC2,[17]Flap structure-specific endonuclease 1,[21]H2AFX,[12]TP53BP1,[12]FANCM,[22]P53,[12][23][24][25]TOP3A,[13][26][27][28]MLH1[18][27][29][30] и CHAF1A[31].
Патофизиология[править | править код]
У больных синдромом Блума наблюдается повышенная частота сестринских хроматидных обменов и, как следствие, повышается шанс возникновения хромосомных разрывов и структурных перестроек хромосом[32]. Детали механизма, связывающего молекулярные процессы, в которые вовлечён белок синдрома Блума и хромосомы, находятся на этапе изучения.
Синдром Блума наследуется по аутосомно-рецессивному механизму. У ребёнка с синдромом Блума оба родителя являются бессимптомными носителями мутации по одному аллелю гена BLM. Частота носителей мутантного гена варьирует в зависимости от популяции. Например, у потомков евреев ашкеназов она составляет 1/100. Синдром проявляется у обоих полов, при этом, однако, у женщин поражение кожи менее сильное, чем у мужчин, поэтому заболевание может быть не диагностировано как синдром Блума. В настоящий момент нет достоверных свидетельств о том, что симптомы синдрома Блума могут варьироваться в зависимости от типа мутации в гене BLM.
Диагностика[править | править код]
Медико-генетическое консультирование и генетический анализ рекомендованы семьям, в которых могут быть носители синдрома Блума. Семьи, в которых статус носителя достоверно известен, могут воспользоваться пренатальным тестированием с применением цитогенетических и молекулярно-биологических методов. Синдром Блума диагностируют при помощи любого из трех тестов: по присутствию так называемых обменных хроматидных аберраций (тетрарадиалов) в культурах лимфоцитов крови, и/или повышенному уровню сестринских хроматидных обменов в клетках любого типа, и/или по наличию мутации в гене BLM[33].
См. также[править | править код]
- Старение (биология)
- Прогерия
Ссылки[править | править код]
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ OMIM 210900
- ↑ James, William; Berger, Timothy; Elston, Dirk. Andrews’ Diseases of the Skin: Clinical Dermatology (англ.). — 10th. — Saunders, 2005. — P. 575. — ISBN 0-7216-2921-0.
- ↑ Karow, JK; Constantinou, A; Li, JL; West, SC; Hickson, I.D. The Bloom’s syndrome gene product promotes branch migration of holliday junctions (англ.) // Proceedings of the National Academy of Sciences of the United States of America : journal. — 2000. — Vol. 97, no. 12. — P. 6504—6508. — doi:10.1073/pnas.100448097. — PMID 10823897.
- ↑ Straughen, Je; Johnson, J; Mclaren, D; Proytcheva, M; Ellis, N; German, J; Groden, J. A rapid method for detecting the predominant Ashkenazi Jewish mutation in the Bloom’s syndrome gene (англ.) // Human Mutation (англ.)русск. : journal. — 1998. — Vol. 11, no. 2. — P. 175—178. — doi:10.1002/(SICI)1098-1004(1998)11:2<175::AID-HUMU11>3.0.CO;2-W. — PMID 9482582.
- ↑ Bloom D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs; probably a syndrome entity (англ.) // A.M.A. American journal of diseases of children : journal. — 1954. — Vol. 88, no. 6. — P. 754—758. — PMID 13206391.
- ↑ Deans A.J., West S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia (англ.) // Molecular Cell (англ.)русск. : journal. — 2009. — December (vol. 36, no. 6). — P. 943—953. — doi:10.1016/j.molcel.2009.12.006. — PMID 20064461.
- ↑ Amor-Guéret M. Bloom syndrome, genomic instability and cancer: the SOS-like hypothesis (англ.) // Cancer Letters (англ.)русск. : journal. — 2006. — Vol. 236, no. 1. — P. 1—12. — doi:10.1016/j.canlet.2005.04.023. — PMID 15950375.
- ↑ German J. Bloom’s syndrome. XX. The first 100 cancers (неопр.) // Cancer Genet Cytogenet. — 1997. — Т. 93, № 1. — С. 100—106. — doi:10.1016/S0165-4608(96)00336-6. — PMID 9062585. (недоступная ссылка)
- ↑ Karow J.K., Chakraverty R.K., Hickson I.D. The Bloom’s syndrome gene product is a 3′-5′ DNA helicase (англ.) // Journal of Biological Chemistry : journal. — 1998. — January (vol. 272, no. 49). — P. 30611—30614. — doi:10.1074/jbc.272.49.30611. — PMID 9388193.
- ↑ Bloom syndrome. Genetics Home Reference. NIH. Дата обращения: 19 марта 2013.
- ↑ 1 2 3 4 Sengupta, Sagar; Robles Ana I., Linke Steven P., Sinogeeva Natasha I., Zhang Ran, Pedeux Remy, Ward Irene M., Celeste Arkady, Nussenzweig André, Chen Junjie, Halazonetis Thanos D., Harris Curtis C. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest (англ.) // Journal of Cell Biology (англ.)русск. : journal. — United States, 2004. — September (vol. 166, no. 6). — P. 801—813. — ISSN 0021-9525. — doi:10.1083/jcb.200405128. — PMID 15364958.
- ↑ 1 2 Brosh, R M; Li J L., Kenny M K., Karow J K., Cooper M P., Kureekattil R P., Hickson I D., Bohr V A. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity (англ.) // Journal of Biological Chemistry : journal. — UNITED STATES, 2000. — August (vol. 275, no. 31). — P. 23500—23508. — ISSN 0021-9258. — doi:10.1074/jbc.M001557200. — PMID 10825162.
- ↑ Opresko, Patricia L; von Kobbe Cayetano, Laine Jean-Philippe, Harrigan Jeanine, Hickson Ian D., Bohr Vilhelm A. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases (англ.) // Journal of Biological Chemistry : journal. — United States, 2002. — October (vol. 277, no. 43). — P. 41110—41119. — ISSN 0021-9258. — doi:10.1074/jbc.M205396200. — PMID 12181313.
- ↑ Moens, Peter B; Kolas Nadine K., Tarsounas Madalena, Marcon Edyta, Cohen Paula E., Spyropoulos Barbara. The time course and chromosomal localization of recombination-related proteins at meiosis in the mouse are compatible with models that can resolve the early DNA-DNA interactions without reciprocal recombination (англ.) // Journal of Cell Science (англ.)русск. : journal. — England: The Company of Biologists (англ.)русск., 2002. — April (vol. 115, no. Pt 8). — P. 1611—1622. — ISSN 0021-9533. — PMID 11950880.
- ↑ von Kobbe, Cayetano; Karmakar Parimal, Dawut Lale, Opresko Patricia, Zeng Xianmin, Brosh Robert M., Hickson Ian D., Bohr Vilhelm A. Colocalization, physical, and functional interaction between Werner and Bloom syndrome proteins (англ.) // Journal of Biological Chemistry : journal. — United States, 2002. — June (vol. 277, no. 24). — P. 22035—22044. — ISSN 0021-9258. — doi:10.1074/jbc.M200914200. — PMID 11919194.
- ↑ 1 2 Braybrooke, Jeremy P; Li Ji-Liang, Wu Leonard, Caple Fiona, Benson Fiona E., Hickson Ian D. Functional interaction between the Bloom’s syndrome helicase and the RAD51 paralog, RAD51L3 (RAD51D) (англ.) // Journal of Biological Chemistry : journal. — United States, 2003. — November (vol. 278, no. 48). — P. 48357—48366. — ISSN 0021-9258. — doi:10.1074/jbc.M308838200. — PMID 12975363.
- ↑ 1 2 Wang, Y; Cortez D., Yazdi P., Neff N., Elledge S J., Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures (англ.) // Genes & Development : journal. — UNITED STATES, 2000. — April (vol. 14, no. 8). — P. 927—939. — ISSN 0890-9369. — PMID 10783165.
- ↑ Beamish, Heather; Kedar Padmini, Kaneko Hideo, Chen Philip, Fukao Toshiyuki, Peng Cheng, Beresten Sergei, Gueven Nuri, Purdie David, Lees-Miller Susan, Ellis Nathan, Kondo Naomi, Lavin Martin F. Functional link between BLM defective in Bloom’s syndrome and the ataxia-telangiectasia-mutated protein, ATM (англ.) // Journal of Biological Chemistry : journal. — United States, 2002. — August (vol. 277, no. 34). — P. 30515—30523. — ISSN 0021-9258. — doi:10.1074/jbc.M203801200. — PMID 12034743.
- ↑ Wu, L; Davies S L., Levitt N C., Hickson I D. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51 (англ.) // Journal of Biological Chemistry : journal. — United States, 2001. — June (vol. 276, no. 22). — P. 19375—19381. — ISSN 0021-9258. — doi:10.1074/jbc.M009471200. — PMID 11278509.
- ↑ Sharma, Sudha; Sommers Joshua A., Wu Leonard, Bohr Vilhelm A., Hickson Ian D., Brosh Robert M. Stimulation of flap endonuclease-1 by the Bloom’s syndrome protein (англ.) // Journal of Biological Chemistry : journal. — United States, 2004. — March (vol. 279, no. 11). — P. 9847—9856. — ISSN 0021-9258. — doi:10.1074/jbc.M309898200. — PMID 14688284.
- ↑ Deans, Andrew; Stephen West. FANCM Connects the Genome Instability Disorders Bloom’s Syndrome and Fanconi Anemia (англ.) // Molecular Cell (англ.)русск. : journal. — 2009. — 24 December (vol. 36, no. 6). — P. 943—953. — doi:10.1016/j.molcel.2009.12.006. — PMID 20064461.
- ↑ Wang, X W; Tseng A., Ellis N A., Spillare E A., Linke S P., Robles A I., Seker H., Yang Q., Hu P., Beresten S., Bemmels N A., Garfield S., Harris C C. Functional interaction of p53 and BLM DNA helicase in apoptosis (англ.) // Journal of Biological Chemistry : journal. — United States, 2001. — August (vol. 276, no. 35). — P. 32948—32955. — ISSN 0021-9258. — doi:10.1074/jbc.M103298200. — PMID 11399766.
- ↑ Garkavtsev, I V; Kley N., Grigorian I A., Gudkov A V. The Bloom syndrome protein interacts and cooperates with p53 in regulation of transcription and cell growth control (англ.) // Oncogene (англ.)русск. : journal. — England, 2001. — December (vol. 20, no. 57). — P. 8276—8280. — ISSN 0950-9232. — doi:10.1038/sj.onc.1205120. — PMID 11781842.
- ↑ Yang, Qin; Zhang Ran, Wang Xin Wei, Spillare Elisa A., Linke Steven P., Subramanian Deepa, Griffith Jack D., Li Ji Liang, Hickson Ian D., Shen Jiang Cheng, Loeb Lawrence A., Mazur Sharlyn J., Appella Ettore, Brosh Robert M., Karmakar Parimal, Bohr Vilhelm A., Harris Curtis C. The processing of Holliday junctions by BLM and WRN helicases is regulated by p53 (англ.) // Journal of Biological Chemistry : journal. — United States, 2002. — August (vol. 277, no. 35). — P. 31980—31987. — ISSN 0021-9258. — doi:10.1074/jbc.M204111200. — PMID 12080066.
- ↑ Wu, L; Davies S L., North P S., Goulaouic H., Riou J F., Turley H., Gatter K C., Hickson I D. The Bloom’s syndrome gene product interacts with topoisomerase III (англ.) // Journal of Biological Chemistry : journal. — UNITED STATES, 2000. — March (vol. 275, no. 13). — P. 9636—9644. — ISSN 0021-9258. — doi:10.1074/jbc.275.13.9636. — PMID 10734115.
- ↑ 1 2 Freire, R; d’Adda Di Fagagna F., Wu L., Pedrazzi G., Stagljar I., Hickson I D., Jackson S P. Cleavage of the Bloom’s syndrome gene product during apoptosis by caspase-3 results in an impaired interaction with topoisomerase IIIα (англ.) // Nucleic Acids Research (англ.)русск. : journal. — England, 2001. — August (vol. 29, no. 15). — P. 3172—3180. — doi:10.1093/nar/29.15.3172. — PMID 11470874.
- ↑ Hu, P; Beresten S F., van Brabant A J., Ye T Z., Pandolfi P P., Johnson F B., Guarente L., Ellis N A. Evidence for BLM and Topoisomerase IIIalpha interaction in genomic stability (англ.) // Human Molecular Genetics (англ.)русск. : journal. — England: Oxford University Press, 2001. — June (vol. 10, no. 12). — P. 1287—1298. — ISSN 0964-6906. — doi:10.1093/hmg/10.12.1287. — PMID 11406610.
- ↑ Langland, G; Kordich J., Creaney J., Goss K H., Lillard-Wetherell K., Bebenek K., Kunkel T A., Groden J. The Bloom’s syndrome protein (BLM) interacts with MLH1 but is not required for DNA mismatch repair (англ.) // Journal of Biological Chemistry : journal. — United States, 2001. — August (vol. 276, no. 32). — P. 30031—30035. — ISSN 0021-9258. — doi:10.1074/jbc.M009664200. — PMID 11325959.
- ↑ Pedrazzi, G; Perrera C., Blaser H., Kuster P., Marra G., Davies S L., Ryu G H., Freire R., Hickson I D., Jiricny J., Stagljar I. Direct association of Bloom’s syndrome gene product with the human mismatch repair protein MLH1 (англ.) // Nucleic Acids Research (англ.)русск. : journal. — England, 2001. — November (vol. 29, no. 21). — P. 4378—4386. — doi:10.1093/nar/29.21.4378. — PMID 11691925.
- ↑ Jiao, Renjie; Bachrati Csanád Z., Pedrazzi Graziella, Kuster Patrick, Petkovic Maja, Li Ji-Liang, Egli Dieter, Hickson Ian D., Stagljar Igor. Physical and Functional Interaction between the Bloom’s Syndrome Gene Product and the Largest Subunit of Chromatin Assembly Factor 1 (англ.) // Molecular and Cellular Biology (англ.)русск. : journal. — United States, 2004. — June (vol. 24, no. 11). — P. 4710—4719. — ISSN 0270-7306. — doi:10.1128/MCB.24.11.4710-4719.2004. — PMID 15143166.
- ↑ German J, Schonberg S, Louie, and Chaganti R S. Bloom’s syndrome. IV. Sister-chromatid exchanges in lymphocytes. Am J Hum Genet. 1977; 29(3): 248—255. PMID 868871
- ↑ Sanz, MM; German, J; Pagon, RA; Adam, MP; Bird, TD; Dolan, CR; Fong, CT; Stephens, K. Bloom’s Syndrome (неопр.) / GeneReviews™ [Internet].Pagon R.A., Adam M.P., Bird T.D., et al. — Seattle, 1993. — PMID 20301572.
Примечания[править | править код]
- Bloom’s Syndrome on GONUTS
- Sanz M. M., German J. Bloom’s Syndrome (неопр.). — 2010. In GeneReviews™ [Internet] (неопр.) / Pagon R. A., Bird T. D., Dolan C. R., et al.. — Seattle WA: University of Washington, Seattle, 1993–.
- Blooms Connect — a forum by and for people with Bloom’s Syndrome and their loved ones
- Bloom’s Syndrome Registry — research and information about Bloom’s Syndrome
- Bloom’s Syndrome Foundation — devoted to medical & scientific research to discover a treatment or cure for Bloom’s Syndrome
- DNA Repair
- Segmental Progeria
- GeneReviews/NCBI/NIH/UW entry on Bloom Syndrome
Источник
Синдром Блума – это редкое врожденное заболевание, при котором клетки человека демонстрируют геномную нестабильность. Наследуется оно по аутосомно-рецессивному типу.
Нарушение впервые диагностировал и описал в 1954 году дерматолог американского происхождения Дэвид Блум. От имени этого ученого и произошло название патологии. Синоним – врожденная телеангиэктатическая эритема.
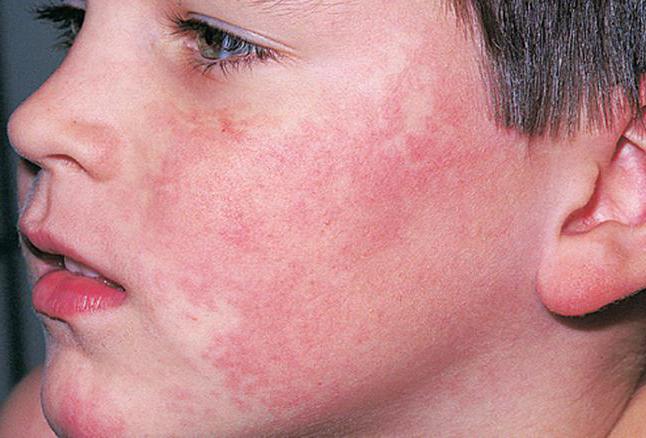
Наиболее часто синдромом Блума страдают лица еврейской национальности (приблизительно 1 человек из 100). Заболевание может быть как у женщин, так и у мужчин, но у последних симптомы более выражены. Именно поэтому женщинам с данной патологией часто ставят ошибочные диагнозы.
У ребенка, который болеет синдромом Блума, оба родителя являются скрытыми носителями мутации по одному аллелю гена BLM. Предполагается, что разнообразие симптомов зависит от того, какая именно мутация присутствует в генах больного. Однако это еще не доказано.
Клиническая картина
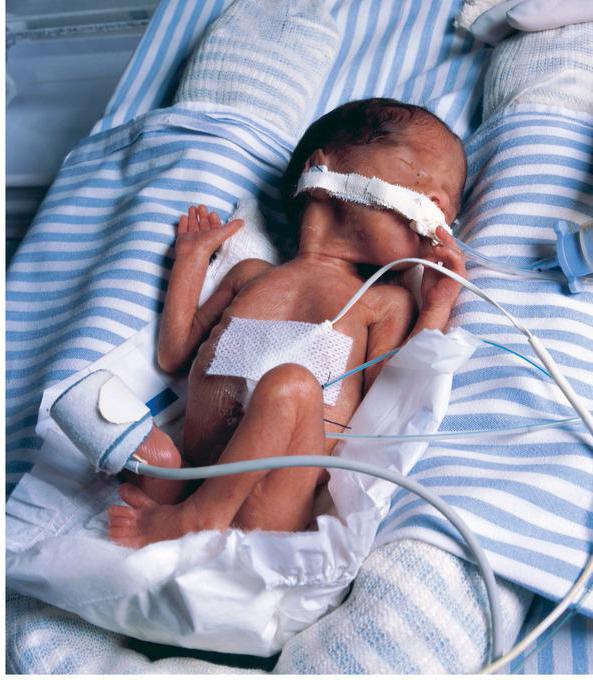
Пациенты с синдромом Блума при рождении имеют небольшую массу тела (около 1900-2000 г). В дальнейшем они также медленно растут и плохо прибавляют в весе. Половое созревание задерживается, и даже если оно проходит, то неполноценно. У мужчин часто встречается бесплодие, а у женщин – аномально ранняя менопауза. Несмотря на это, их психическое развитие соответствует возрастным нормам.
В первые недели жизни у больных на щеках, ушах, носу и тыльной части кистей рук возникают пузыри, эритема и корки. Нередко отмечается повышенная чувствительность к ультрафиолету. Даже непродолжительное нахождение пациентов под солнцем может привести к образованию сосудистой сетки и повреждениям кожных покровов различной степени тяжести. После восстановления облученной кожи на ней могут образоваться темные или слишком светлые пятна, участки с атрофией.
Больные имеют сниженный иммунитет и поэтому часто сталкиваются с инфекционными заболеваниями, которые к тому же рецидивируют.
Синдром Блума часто сочетается с асимметрией шеек костей бедра и врожденными пороками сердца.
Внешний вид
Внешний вид больных нестандартный. У них достаточно узкий череп, маленький подбородок и выступающий нос («птичье лицо»). Особенно хорошо это видно, если посмотреть фото синдрома Блума.
Больные, как правило, низкого роста, имеют высокий голос. У некоторых пациентов присутствуют деформации стоп и аномалии зубов. Больные нередко жалуются на воспаление и отечность губ, их шелушение. В некоторых случаях отмечается нарушение процесса ороговения кожи и ее закупорка (выглядит как «гусиная кожа»).
Диагностика
Диагноз “синдром Блума” ставит врач, опираясь на клиническую картину болезни пациента и данные результатов лабораторных исследований.
При обследовании обязательно проводится оценка состояния иммунной системы. В анализе больных синдромом Блума будет отмечаться снижение количества иммуноглобулинов и Т-лимфоцитов. Кроме этого, рекомендуется провести оценку сестринских хроматидных обменов.
При диагностике важно не спутать синдром Блума с красной волчанкой, синдромом Нила-Дингуолла, синдромом Ротмунда-Томсона и кожной порфирией.
Онкология
Низкий иммунитет и наличие большого количества различных мутаций приводят к тому, что у больного резко возрастает вероятность появления онкологии. Страдать при этом могут как внутренние органы, так и кровь, лимфа, костная ткань.
К самым распространенным патологиям, возникающим у этой категории пациентов, можно отнести:
- миелоидный лейкоз;
- лимфому;
- лимфолейкоз;
- злокачественные опухоли пищевода, языка и кишечника;
- рак легких;
- карциному молочных желез.
Гораздо реже у них диагностируется медуллобластома и рак почки.
Лечение
Пациент, страдающий синдромом Блума, лечение будет проходить симптоматическое. Уменьшения выраженности неприятных явлений можно добиться при помощи медикаментозных средств и лечебных процедур. Их выбор осуществляется лечащим врачом исходя из того, какие симптомы тревожат больного. Так, при онкологии может потребоваться химиотерапия, лучевая терапия или хирургическая операция, при болезнях зубов – стоматологические процедуры и т. д. Излечиться полностью от патологии на данный момент невозможно.
В любом случае больные должны регулярно применять средства, которые надежно защищают кожу от ультрафиолетового излучения, употреблять витаминно-минеральные комплексы (в них должен входить витамин Е), каротиноиды (как в форме пищевых добавок, так и с пищевыми продуктами) и лекарства, корректирующие работу иммунной системы. В некоторых случаях возможно проведение гормональной терапии.

Пациенты, у которых синдром Блума, на протяжении всей жизни должны наблюдаться у дерматолога и онколога, знать первые признаки рака кожи. При любых подозрительных изменениях им нужно в самые короткие сроки обратиться к врачу.
Больным, на теле которых много родимых пятен, полезно находиться в тени и избегать прямых солнечных лучей, носить максимально скрывающую тело одежду.
Прогноз
Прогноз пациентов с синдромом Блума напрямую зависит от того, какие патологические состояния их сопровождают. Наиболее часто больные умирают в результате острых онкологических процессов или пневмонии.
Стоит отметить, что пациенты, которые проходят симптоматическое лечение и находятся под наблюдением врача, имеют большую продолжительность жизни в сравнении с теми больными, которые этого не делают.
Профилактика
Профилактика синдрома Блума у детей будет состоять в избегании браков с близкими родственниками. Именно у народов, в традиции которых близкородственные браки, болезнь находит наиболее частое распространение.
Кроме этого, молодой паре перед зачатием рекомендуется пройти тщательное медицинское обследование.
Источник
Что такое синдром Блума?
Синдром Блума, также называемый синдромом Блума-Торре-Мачэйкик или врожденной телеангиэктатической эритемой, представляет собой редкий генодерматоз, характеризующийся нестабильностью генома и предрасположенностью к развитию различных видов рака. Синдром Блума вызван мутациями в гене BLM, который индуцирует образование аномального белка ДНК-геликазы. Наиболее заметные признаки включают недостаточность роста в пренатальном периоде, легкий иммунодефицит, чрезмерную светочувствительность с кожными поражениями, напоминающими волчанку на лице, сахарный диабет 2 типа и гипогонадизм. Повышенный риск злокачественных новообразований при синдроме Блума приводит к сокращению продолжительности жизни и увеличению патологической поражённости у больных.
Признаки и симптомы

Наиболее постоянным клиническим признаком синдрома Блума, наблюдаемым на всех этапах жизни, является плохой рост, который влияет на рост, вес и окружность головы. Этот дефицит роста начинается до рождения, и пораженный плод обычно меньше нормального для гестационного возраста. Средний вес при рождении больных мальчиков составляет 1760 г (диапазон 900–3189 г), а девочек — 1754 г (диапазон 700–2892 г). Тем не менее пропорции тела нормальные. Средний рост взрослых больных мужчин и женщин составляет 149 см (диапазон 128–164 см) и 138 см (диапазон 115–160 см), соответственно.
Хотя внешность людей с расстройством различна и может быть неотличима от здоровых людей того же возраста и размера, младенцы и взрослые с синдромом Блума обычно имеют отчетливо узкую голову и лицо, но при этом имеют нормальные пропорции тела. Из-за разреженного подкожного жира нос и/или уши могут выступать. Несмотря на очень маленькую окружность головы, большинство больных имеют нормальные интеллектуальные способности.
Более подробно о симптомах написано ниже в разделе «История и физика».
Причины
Синдром Блума — редкое аутосомно-рецессивное заболевание хромосомной нестабильности. Расстройство связано с мутациями в гене BLM, расположенном в 15q26.1. Ген BLM кодирует BLM геликазу, которая образует комплекс с двумя другими белками, ДНК-топоизомеразой IIIα и RMI. Мутации в гене BLM вызывают ошибки во время репликации ДНК и приводят к более многочисленным хромосомным перестройкам и поломкам. В результате высокой степени нестабильности генома люди с синдромом Блума подвергаются повышенному риску развития злокачественных новообразований.
Эпидемиология
Синдром Блума — крайне редкое заболевание для большинства групп населения, по состоянию на 2018 год в реестре синдромов Блума числился только 281 пациент. Чаще всего он описывался среди еврейского населения ашкенази. На их долю приходится 25% пораженных людей во всем мире (примерно 1% евреев ашкенази являются носителями мутации BLMAsh). В Соединенных Штатах Америки зарегистрировано около 170 случаев. У детей чьи родители кровные родственники расстройство встречается чаще, чем среди населения в целом. Есть небольшое преобладание мужчин.
Патофизиология
Ген BLM кодирует геликазу RecQ, RECQL3, иначе известную как белок синдрома Блума. Геликазы раскручивают двухцепочечную и многоцепочечную ДНК или РНК во время репликации и транскрипции. В частности, семейство геликаз RECQ действует как хранитель генома, поддерживая стабильность ДНК в ответ на репликативный, транскрипционный и теломерный стресс. Это семейство геликаз высоко консервативно у всех видов, у млекопитающих существует пять RECQ-подобных (RECQL) геликаз. Из пяти геликаз RECQL три были связаны с прогероидными и/или канцерогенными заболеваниями у людей. К ним относятся синдром Блума, синдром Вернера и синдром Ротмунда-Томсона с мутациями в BLM, WRN и RECQL4 соответственно. Мутировавшие геликазы RECQL обладают вредоносными геномными эффектами из-за нарушения регуляции раскручивания ДНК. В частности, у пациентов с синдромом Блума скорость сестринских хроматидных обменов увеличивается в 10 раз. Кроме того, преобладание хромосомных разрывов и перестроек при синдроме Блума дополнительно подчеркивает важную функцию геликаз RECQL в регуляции генома.
Гистопатология
Гистопатология при синдроме Блума часто имитирует красную волчанку. Отмечается выраженная вакуолярная граница с дегенерацией базального разжижения, стиранием сетчатых гребней и закупоркой фолликулов. Утолщение базальной мембраны может быть замечено, а может и нет. В дерме имеется умеренный поверхностный инфильтрат лимфоцитов, преимущественно Т-лимфоцитов. Расширение капилляров также присутствует в сосочковом слое дермы. Прямая иммунофлуоресценция показывает неспецифические отложения IgM вдоль зоны базальной мембраны пораженной кожи.
Диагностика
— История и физика.
Лица с синдромом Блума отличаются тяжелой пренатальной и послеродовой задержкой роста, микроцефалией и характерными чертами лица (килевидная форма, гипоплазия скуловой кости, выступающие носы и выпуклые уши). Также часто присутствуют высокий голос и отсутствие верхних боковых резцов. Родители детей с синдромом Блума часто обращаются за медицинской помощью из-за небольшого роста ребенка при пропорциональном размере тела. Таким образом, этим детям часто ошибочно ставят диагноз карликовость.
Кожные особенности включают светочувствительность и характерную сыпь на скулах, напоминающую волчанку. Эритематозные поражения, вызванные УФ-излучением, могут аналогичным образом возникать на других участках, подверженных воздействию солнечного света, таких как предплечья и тыльная сторона кистей. Телеангиэктазии и пойкилодермия возникают в начале первого или второго года жизни в ответ на воздействие солнца. Небольшие скопления телеангиэктазов часто появляются внутри сыпи и склер глаз. Другие кожные признаки включают трещины во рту, пятна с молоком и хорошо разграниченные очаги дисхромии (гипо- и гиперпигментации).
К другим аномалиям относятся гипогонадизм, мужское бесплодие, преждевременная менопауза у женщин, умственная отсталость и инсулиннезависимый сахарный диабет с поздним началом. Пациенты имеют легкий иммунодефицит, приводящий к рецидивирующему среднему отиту и синопульмональным инфекциям в младенчестве. Это происходит из-за пониженных уровней иммуноглобулинов плазмы, включая IgA, IgM и иногда IgG. В младенчестве из-за рвоты, диареи и гастроэзофагеального рефлюкса может возникнуть сильное обезвоживание. Аспирация при рефлюксной болезни может способствовать увеличению заболеваемости пневмонией и развитию хронических заболеваний легких у этих пациентов. Легочная недостаточность — вторая ведущая причина смерти в этой популяции.
Пациенты с синдромом Блума имеют высокую предрасположенность к различным злокачественным новообразованиям. Почти у 50% пациентов с синдромом Блума в какой-то момент жизни будет диагностирован рак. Развитие опухоли происходит в среднем в более раннем возрасте, чем у населения в целом. Пациенты могут иметь множественные независимые первичные раковые опухоли различных типов и локализации. Средний возраст диагноза рака составляет 23 года, и смерть обычно наступает до 30 лет. Лейкемии (миелоидные или лимфоидные) и неходкинские лимфомы являются наиболее распространенным типом рака в первые два десятилетия жизни (средний возраст 20 лет). Лимфомы возникают в 150–300 раз чаще, чем у здорового населения. К сожалению, у 10% пациентов разовьется второе злокачественное новообразование. К ним могут относиться рак легких, желудочно-кишечного тракта (особенно колоректальный рак), груди, печени, кожи (базальноклеточная карцинома и плоскоклеточный рак), остеосаркома, ретинобластома и опухоли головного мозга. Осложнения злокачественных новообразований — основная причина смерти.
— Анализы.
При наличии клинического подозрения диагноз синдрома Блума подтверждается цитогенетическим анализом, показывающим повышенное количество сестринских хроматидных обменов или квадрирадиальных конфигураций в лимфоцитах или фибробластах. Также может быть выполнен целевой мутационный анализ. Для популяций носителей высокого риска рекомендуется генетическое и пренатальное тестирование.
Лабораторные исследования включают полный анализ крови с дифференциальным показателем низких нормальных лимфоцитов. Эти пациенты часто имеют более высокие эффекторные Т-клетки памяти, но пониженные В-клетки памяти. Уровни IgM, IgA и иногда IgG в плазме крови снижаются.
Лечение
В ведении таких пациентов особенно важен мультидисциплинарный подход. Из-за редкости этого состояния и его сложности нет единого мнения по поводу ведения или лечения. В неонатальном периоде необходимо строго контролировать потребление жидкости, чтобы предотвратить опасное для жизни обезвоживание. Дополнительное питание через гастростомические зонды может быть полезно для предотвращения обезвоживания и недоедания, но, по-видимому, не улучшает линейный рост. Введение гормона роста кажется неэффективным для увеличения скорости роста или роста взрослого человека. Кроме того, это может увеличить риск развития опухоли. Для лечения инфекций используются соответствующие антибиотики. Заместительная терапия иммуноглобулином может использоваться у пациентов со значительным иммунодефицитом. Для лечения сахарного диабета необходимо тщательное наблюдение эндокринолога.
Согласно одному исследованию, следует избегать гематологического скрининга на злокачественные новообразования у детей из-за отсутствия прогностических преимуществ при ранней диагностике. Однако взрослым полезно хирургическое удаление карцином на ранней стадии, поэтому рекомендуется ежегодно проходить обследование на рак груди, шейки матки и толстой кишки. Из-за высокого риска хромосомного разрыва необходимо минимизировать радиационное воздействие. Для скрининга и диагностики рака вместо компьютерной томографии и рентгенографии следует использовать магнитно-резонансную томографию и ультразвук. Из-за риска повышенной токсичности химиотерапевтические агенты могут нуждаться в корректировке дозы. В частности, было показано, что 5-фторурацил вызывает более высокие уровни фрагментации ДНК. Следует избегать лучевой терапии. Было показано, что протонная лучевая терапия является более безопасной альтернативой лучевой терапии.
Прогноз
Многие пациенты с синдромом Блума доживают до зрелого возраста. Средний возраст смерти — 26 лет, чаще всего от осложнений злокачественного новообразования. Вторая по частоте причина смерти — хронические заболевания легких.
Осложнения
У пациентов часто развивается серьезное угнетение костного мозга и токсичность даже при уменьшенных дозах химиотерапии и облучения. Следовательно, злокачественные новообразования очень трудно лечить; хотя протонная лучевая терапия добилась определенных успехов. Однако даже пациенты в стадии ремиссии рака часто умирают от осложнений, вызванных пневмонией, хроническим заболеванием легких, заболеванием печени или сепсисом.
Список источников:
https://www.ncbi.nlm.nih.gov/books/NBK448138/
https:/rarediseases.org/rare-diseases/bloom-syndrome/
Источник