
Синдром чедиака хигаси является следствием поражения


Синдром Чедиака-Хигаси — редкое аутосомно-рецессивное заболевание, которое характеризуется тяжелым врожденным иммунодефицитом по причине дефекта гена CHS1/LYST. Проявляется болезнь частыми бактериальными инфекциями, коагулопатией, нарушениями пигментации кожи, а также прогрессирующими неврологическими расстройствами [1].
Иммунологические нарушения выражаются в нейтропении и нехватке естественных киллеров (NK-клеток). Зачастую заболевание оказывается летальным — большая часть пациентов умирает в детстве. Заподозрить синдром Чедиака-Хигаси можно по тем же признакам, что и любой врожденный иммунодефицит, однако имеется и довольно специфичный критерий — гигантские внутриклеточные включения (или органеллы) в популяциях разных клеток и определенные дефекты в них [2].
Чтобы более наглядно описать происходящие процессы при данном заболевании, еще во второй половине прошлого столетия была предложена оригинальная модель «бежевой мыши». Вследствие мутации в определенном гене (в то время конкретная точка генома, разумеется, не была известна) пигментация экспериментальной мыши изменялась, и исследователи определили ее цвет как «бежевый» («beige»). Помимо цвета у таких животных имелся еще ряд аномалий в виде огромных гранул меланина в лимфоцитах, нейтрофилах и эозинофилах (рис. 1), также подобные включения наблюдались в ряде других клеток [3].
Рисунок 1 | Периферическая кровь бежевой мыши. Световая микроскопия: определяются гранулы в цитоплазме лимфоцита (А) и нейтрофила (В). Электронная микроскопия: объемная гранула с темным содержимым в цитоплазме лимфоцита. Относится к непостоянным аномальным включениям [3].
Позднее данный ген у мыши был идентифицирован и назван Lyst (кодирует регулятор лизосомального транспорта — lysosomal trafficking regulator), человеческий ген обозначают как LYST. Фермент, за экспрессию которого отвечает LYST, обеспечивает нормальный экзо- и эндоцитоз лизосомы; в случае мутации лизосома не может выполнять свои функции [4].
Сегодня этот ген настолько прочно ассоциируется с синдромом Чедиака-Хигаси, что в современной литературе его обозначают как CHS1/LYST(человеческий ген Chediak-Higashi syndrome-1 или LYST — оба варианта равнозначны) [1].
Роль регулятора лизосомального транспорта (LYST)
Данный белок кодируется достаточно объемным геном LYST, который содержит 55 экзонов и расположен в локусе 1q43. Большая длина нуклеотидной последовательности этого гена — фактор неоднозначный: с одной стороны, это существенно затрудняет диагностику, с другой — большое количество мутаций вообще никак не влияет на жизнедеятельность человека и не вызывает синдром Чедиака-Хигаси [5].
Ген кодирует одноименный большой каталитический белок — регулятор лизосомального транспорта (425 kDa, 3801 аминокислотное основание). Протеин относится к семейству BEACH — (Beige and Chediak-Higashi) — и, по-видимому, стал первым его представителем [4]. Белки данного семейства имеют три общих С-концевых домена: PH (Pleckstrin-homology домен), BEACH (Beige and Chediak-Higashi домен) и WD40-повторения (рис. 2) [5].
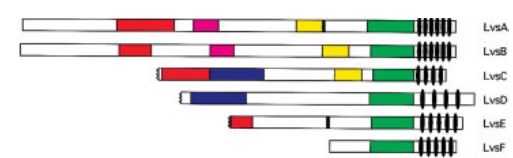
Рисунок 2 | Строение шести родственных белков семейства BEACH слизевика рода Dictyostelium [6].
На рис. 2 изображена диаграмма, наглядно показывающая общность строения шести белков семейства BEACH (в данном случае — шесть белков слизевика рода Dictyostelium). Зеленым цветом обозначен собственно BEACH-домен, наиболее консервативный во всех белках. «Овалами» у С-конца обозначены WD40-повторения — участки, предположительно вовлеченные в белок-белковые взаимодействия. Другими цветами показаны иные идентичные аминокислотные последовательности [6].
Сегодня известно, что белки данного семейства есть у разных представителей животного мира. Имея примерно одинаковое строение, они выполняют многочисленные и крайне сложные функции, обсуждение которых выходит за рамки данной темы.
Собственно LYST обеспечивает секрецию лизосомальных ферментов. Без белка-регулятора транспорт протеолитических ферментов невозможен, вследствие этого цитотоксическая активность фагоцитарных клеток нарушается. Этим также объясняется нарушение транспорта различных метаболитов (накопление в клетках гранул с меланином, что обеспечивает яркий клинический признак синдрома Чедиака-Хигаси), а также нарушения метаболизма некоторых других белков, что обусловливает клиническую картину заболевания и сопутствующие патологии, точный механизм развития которых все еще неизвестен [1,3].
Стоит также заметить, что в последнее время появляются сообщения о преимущественном влиянии дефекта LYST именно на NK-клетки. По последним данным, в лимфоцитах происходит нарушение экзоцитоза протеолитических ферментов, хотя цитокиновая регуляция остается в норме [7].
Клиническая картина
Существует два основных клинических проявления синдрома Чедиака-Хигаси: «парциальный альбинизм» и рецидивирующие пиогенные инфекции. Оба, на первый взгляд, не связанных между собой критерия имеют единую причину — дефект LYST, вследствие чего нарушается транспорт меланина и лизосомальных ферментов.
Цвет волос пациента может быть от сероватого до буквально седого — зависит от этнической принадлежности пациента. Также нарушается пигментация радужной оболочки, следствием чего становится фоточувствительность и сниженная функция зрительного аппарата.
Рецидивирующие бактериальные (в том числе оппортунистические) инфекции часто поражают дыхательные пути, кожу. Возможны дефекты свертывающей системы из-за нарушенного синтеза тромбоцитов, проявляется это небольшими кровоподтеками (зачастую — на слизистых), однако иногда развивается полноценный геморрагический синдром.
Помимо этого возможны неврологические нарушения: атаксия, сенсорные расстройства, прогрессирующая нейродегенерация [1].
Первые проявления заболевания начинаются с раннего детства. Наиболее опасными осложнениями считаются инфекции, для предупреждения которых допустима профилактическая антибиотикотерапия. И все же большинство пациентов умирает в первые десять лет жизни. Наиболее распространенными причинами смерти являются кровотечения, инфекции или гемофагоцитарный лимфогистиоцитоз [7].
Гемофагоцитарный лимфогистиоцитоз
Гемофагоцитарный лимфогистиоцитоз (ГФЛГ) — патологическое состояние, которое может быть вызвано разнообразными причинами, часто — первичными иммунодефицитами (к числу которых относится и синдром Чедиака-Хигаси). Суть данного состояния — гиперпродукция гистиоцитов и иммунокомпетентных клеток [5].
Как уже было сказано выше, мутация LYST нарушает цитотоксическую функцию клеток, но не метаболизм регуляторных факторов. В ответ на антиген развивается обыкновенная воспалительная реакция — однако элиминация чужеродного агента невозможна. В связи с этим продолжается продукция провоспалительных факторов (ИФН-γ, ФНО, интерлейкины), что увеличивает количество лимфоцитов, нейтрофилов, повышает активность макрофагов. Гиперпродукция цитокинов, не ингибированная по механизму обратной связи, иногда называется «цитокиновым штормом» [5].
Следствием этого является лимфогистиоцитарная инфильтрация различных тканей с развитием в них разнообразных повреждений, а макрофаги могут разрушать нормальные функционирующие клетки (в том числе — форменные элементы крови) [5].
Диагностика синдрома Чедиака-Хигаси и сопутствующих заболеваний
В диагностировании синдрома Чедиака-Хигаси особых сложностей не возникает. Как уже было сказано выше, основная задача клинициста — распознать врожденное иммунодефицитное состояние, дальнейшая диагностика основана на данных иммунограммы и генетического анализа. Достаточно специфичным признаком является накопление больших внутриклеточных везикул в различных клетках, в том числе — лейкоцитах [2].
Одним из наиболее серьезных осложнений при данном синдроме является развитие ГФЛГ. Диагностические критерии при этом патологическом состоянии можно представить следующим образом [5]:
- Наличие генетического дефекта, связанного с ГФЛГ (зачастую это первичные иммунодефициты).
- Наличие как минимум пяти из нижеперечисленных критериев:
- лихорадка;
- спленомегалия;
- цитопения хотя бы в двух клеточных популяциях:
а) гемоглобин < 90 г/л (для новорожденных — < 100 г/л);
б) тромбоциты < 100×109/л;
в) нейтрофилы < 1×109/л;
- гипертриглицеридемия (> 3 ммоль/л) или гипофибриногенемия (< 1,5 г/л);
- гиперферритинемия > 500 мкг/л;
- растворимые молекулы CD25 > 2400 Ед/мл;
- гемофагоцитоз в костном мозге, селезенке, лимфоузлах
- низкая (вплоть до полного отсутствия) цитотоксическая активность NK-клеток.
Важно отметить: ГФЛГ является настолько частым серьезным осложнением, что в современной литературе принято разделять синдром Чедиака-Хигаси на две формы: «классическую» (с развитием ГФЛГ) и «атипичную» (без такового) [5].
Лечение синдрома Чедиака-Хигаси и сопутствующих патологий
Как и в большинстве наследственных иммунодефицитов, вариантов терапии немного. Наиболее распространенным методом лечения врожденных иммунологических нарушений является пересадка гемопоэтических клеток; данный синдром не является исключением.
В 2007 году в «Bone Marrow Transplantation» появилась публикация, авторы которой сообщали о 35 случаях проведения пересадки гемопоэтических клеток пациентам с синдромом Чедиака-Хигаси [8]. Перед проведением операции лечение осуществлялось в лучшем случае патогенетическое, призванное замедлить прогрессирование заболевания, избежать осложнений и подготовить пациента к пересадке гемопоэтических клеток.
Всего 13 пациентов получили материал от HLA-идентичного донора (родного брата или сестры), 10 — от родственника, еще 12 — от несвязанного донора. По результатам проведенной терапии, 27 (77 %) из 35 пациентов достигли ремиссии, однако 5-летняя выживаемость составила 62 % (22 пациента).
Наиболее распространенными причинами смерти были посттрансплантационные осложнения и хронические заболевания. Среди умерших — большинство получило аллотрансплантат от не полностью подходящего донора.
Таким образом, исследователи пришли к выводу, что подобная методика может быть достаточно эффективной, если донор и реципиент являются родственниками. Также следует отметить, что шесть пациентов на момент проведения операции были старше девяти лет, что свидетельствует о существенных успехах в сдерживании и предотвращении развития осложнений у пациентов (одному из реципиентов на момент трансплантации было 19 лет).
На современном этапе пересадка гемопоэтических клеток является наиболее эффективным способом лечения синдрома Чедиака-Хигаси [9]. Вероятно, в будущем станет возможна коррекция непосредственно LYST с помощью генно-инженерных методик (например, подобная терапия уже существует для хронической гранулематозной болезни — другого наследственного иммунодефицитного состояния [10]), однако пока что такой альтернативы для синдрома Чедиака-Хигаси нет.
Часто синдром Чедиака-Хигаси осложняется ГФЛГ — тяжелым состоянием, которое требует немедленного лечения. Среди наиболее эффективных методов терапии в гайдлайне 2004 года описаны химиотерапия и пересадка гемопоэтических клеток [11].
Также на протяжении лечения, до и после него в профилактических целях используется антибиотикотерапия [1,11].
Источники:
- J. Kaplan, I. De Domenico, and D. M. Ward, ‘Chediak-Higashi syndrome’, Curr. Opin. Hematol., vol. 15, pp. 22–29, 2008.
- D. L. Nagle et al., ‘Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome’, Nat. Genet., vol. 14, no. 3, pp. 307–311, 1996.
- M. A. Lutzner, C. T. Lowrie, and H. W. Jordan, ‘Giant granules in leukocytes of the beige mouse’, J. Hered., vol. 58, no. 6, pp. 299–300, 1967.
- M. D. F. S. Barbosa et al., ‘Identification of the homologus beige and Chediak-Higashi syndrome genes’, Nature, vol. 382, pp. 262–265, 1996.
- S. Ehl and G. de Saint Basile, ‘Genetic Diseases Predisposing to HLH’, Stiehm’s Immune Defic., pp. 437–460, 2014.
- N. Wang, W. I. Wu, and A. De Lozanne, ‘BEACH family of proteins: Phylogenetic and functional analysis of six Dictyostelium BEACH proteins’, J. Cell. Biochem., vol. 86, no. 3, pp. 561–570, 2002.
- A. Gil-Krzewska et al., ‘Chediak-Higashi syndrome: Lysosomal trafficking regulator domains regulate exocytosis of lytic granules but not cytokine secretion by natural killer cells’, J. Allergy Clin. Immunol., vol. 137, no. 4, pp. 1165–1177, 2016.
- M. Eapen et al., ‘Hematopoietic cell transplantation for Chediak-Higashi syndrome’, Bone Marrow Transplant., vol. 39, no. 7, pp. 411–415, 2007.
- M. L. Lozano, J. Rivera, I. Sánchez-Guiu, and V. Vicente, ‘Towards the targeted management of Chediak-Higashi syndrome’, Orphanet J. Rare Dis., vol. 9, no. 1, 2014.
- M. G. Ott et al., ‘Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1’, Nat. Med., vol. 12, no. 4, pp. 401–409, 2006.
- Jan-Inge Henter, ‘HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocutic Lymphohistiocytosis’, Pediatr. Blood Cancer, vol. 48, pp. 124–131, 2007.
Нашли опечатку? Выделите фрагмент и нажмите Ctrl+Enter.
Источник
Синдром Чедиака-Хигаси. Клиника и диагностика
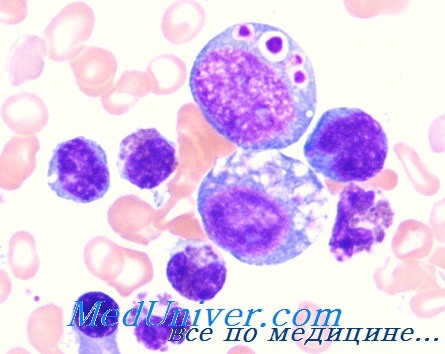
При редком аутосомно-рецессивном синдроме Чедиака-Хигаси повышенная восприимчивость к инфекциям связана с нарушением дегрануляции нейтрофилов. Характерна легкая кровоточивость, депигментация кожи и глаз, прогрессирующая периферическая невропатия и гиперплазия лимфоидной ткани. В нейтрофилах, моноцитах и лимфоцитах обнаруживаются гигантские цитоплазматические гранулы. Депигментация волос, кожи и глазного дна обусловлена патологической агрегацией меланосом и связана с нарушением перекреста зрительных и слуховых нервов. Повышенная восприимчивость к инфекциям частично объясняется нарушениями хемотаксиса, дегрануляции и бактерицидной активности нейтрофилов.
Гигантские гранулы в них препятствуют прохождению клеток в ткани через узкие межклеточные промежутки эндотелия.
Генетика и патогенез синдрома Чедиака-Хигаси. Мутантный ген, расположенный на хромосоме 1 (участок q2-q44), клонирован. Он гомологичен гену, ответственному за соответствующий синдром у мышей (фенотип beige); его продукт обладает структурным сходством с дрожжевым белком сортировки вакуолей, носящим название VPS15. Считается, что этот белок участвует в транспорте везикул и опосредует межбелковые взаимодействия и связь белков с мембранами.
Почти все клетки больных с синдромом Чедиака-Хигаси содержат крупные лизосомы неправильного строения, плотные гранулы или везикулярные структуры. Из-за нарушения дисперсии гигантских меланосом они не поступают в кератиноциты и волосяные фолликулы, вследствие чего стержни волос лишаются пигментных гранул. В результате волосы и кожа больных выглядят светлее, чем у родителей. Те же нарушения меланоцитов обусловливают частичный глазной альбинизм, сопровождающийся светобоязнью.
Слияние гигантских первичных гранул друг с другом и с компонентами цитоплазматических мембран на ранних стадиях развития нейтрофилов приводит к образованию огромных вторичных лизосом с уменьшенным содержанием гидролитических ферментов — протеаз, эластазы и катепсина G. Это и может лежать в основе нарушенной бактерицидной активности нейтрофилов. Повышенная текучесть клеточных мембран при синдроме Чедиака-Хигаси сказывается на экспрессии рецепторов, приводя к повышению внутриклеточного уровня цАМФ, нарушению сборки микротрубочек и их взаимодействия с мембранами лизосом.
Клинические проявления синдрома Чедиака-Хигаси. Кожа у больных светлая, волосы имеют серебристый оттенок. Нередко отмечаются солнечные ожоги и светобоязнь. Характерны частые инфекции и невропатия. Инфекции поражают слизистые оболочки, кожу и дыхательные пути. Повышена восприимчивость как к грамположительным, так и грамотрицательным бактериям и грибам. Наиболее частый возбудитель S. aureus. Невропатия бывает сенсорной и моторной, приводя к атаксии. Нередко она развивается в подростковом возрасте и становится основной жалобой.
Время кровотечения увеличено; количество тромбоцитов нормально, но их агрегация (из-за дефицита плотных гранул, содержащих АДФ и серотонин) нарушена. Нарушена и функция NK-клеток.
Наиболее опасное осложнение синдрома Чедиака-Хигаси — ускорение пролиферации лимфоцитов с панцитопенией, высокой температурой тела и лимфогистиоцитарной инфильтрацией печени, селезенки и лимфатических узлов. Это может произойти в любом возрасте и, по-видимому, связано с отсутствием сопротивляемости вирусу Эпштейна-Барр. Клиническая картина в таких случаях напоминает вирусный гемофагоцитарный синдром. Пролиферация лимфоцитов сопровождается учащением бактериальных и вирусных инфекций и обычно приводит к смерти. Многочисленные лимфогистиоцитарные инфильтраты, обнаруживаемые при аутопсии в печени, селезенке и лимфатических узлах, лишены гистологических признаков злокачественности.
Лабораторные исследования синдрома Чедиака-Хигаси. Диагностическим признаком синдрома Чедиака-Хигаси служат крупные включения во всех ядерных клетках крови. Они видны при окраске мазков крови по Райту, но лучше выявляются на пероксидазе.
Лечение синдрома Чедиака-Хигаси. Некоторым больным в стабильной фазе синдрома помогают высокие дозы аскорбиновой кислоты (200 мг/сут для грудных детей и 2000 мг/сут для взрослых). Ее эффективность вызывает сомнения, но безопасность витамина С оправдывает его введение всем больным.
Единственно надежный метод лечения синдрома Чедиака-Хигаси при ускоренной пролиферации лимфоцитов — трансплантация костного мозга от HLA-идентичных родственников или доноров, совместимых по локусу D. Трансплантация костного мозга в таких случаях нормализует гемопоэтические и иммунологические функции и восполняет дефицит NK-клеток, но не излечивает и не предотвращает невропатию.
– Также рекомендуем “Недостаточность миелопероксидазы (МПО). Клиника и диагностика”
Оглавление темы “Нейтропении”:
- Синдром Чедиака-Хигаси. Клиника и диагностика
- Недостаточность миелопероксидазы (МПО). Клиника и диагностика
- Хроническая гранулематозная болезнь. Причина и клиника
- Диагностика и лечение хронической гранулематозной болезни
- Нейтропения и ее причины. Лекарственная нейтропения
- Иммунная нейтропения детей и новорожденных
- Наследственная нейтропения. Тяжелая врожденная нейтропения – синдром Костманна
- Синдром Швахмана-Даймонда. Метафизариая хондродисплазия тип Мак-Кьюсика и гликогеноз
- Клиника и диагностика нейтропении
- Лечение нейтропении. Терапия хронической врожденной нейтропении у детей
Источник
Под синдромом Чедиака-Хигаси понимается генетически обусловленное патологическое заболевание, которое развивается и прогрессирует в детском возрасте. Клиническая картина течения болезни неблагоприятная, так как у пациентов с синдромом появляется склонность к повышенному риску вирусного или бактериального инфицирования.

Патогенез синдрома Чедиака-Хигаси
Патогенез связан с аномалией в структуре клеточных мембран, сбоем в системе собирательных микротрубочек и нарушением взаимодействия с мембранами лизосом. Клинические проявления можно объяснить неправильным распределением лизосомальных ферментов. На частоту и тяжесть пиогенных инфекций влияет снижение активности кислородного метаболизма и внутриклеточное переваривание микробов в фагоцитах по причине отставания и неполного высвобождения из гигантских гранул в фагосомы гидролитических лизосомальных ферментов. У больных снижается антителозависимая цитотоксичность лимфоцитов и активность естественных клеток-киллеров. Патология относится к первичному иммунодефициту.
В чем опасность?
Опасность синдрома кроется в склонности организма под влиянием патологии формировать вредоносные новообразования, а именно раковые клетки и злокачественные опухоли. Чаще всего такие дети погибают в возрасте до 10 лет, то есть не доживают до подросткового периода. Синдром Чедиака-Хигаси в иммунологии встречается не слишком часто. Он наследуется по аутосомно-рецессивному типу.

Характер болезни
Заболевание носит наследственный характер, однако прогрессирует по типу аутосомно-рецессивной патологии и, как правило, ей сопутствует нарушение пигментации, а также повышенная чувствительность к различным вредоносным микроорганизмам по причине выведения из строя нейтрофилов. Именно в них находятся аномальные клетки, обладающие лизосомными ферментами, кислыми фосфатазами и пероксидазой.
Ведь синдром Чедиака-Хигаси является следствием поражения клеточных мембран. Подобные патогенные образования находятся преимущественно в лимфоцитах, эритроцитах, тромбоцитах, а также кожных фибробластах, впоследствии из-за активного роста могут вызывать такие серьезные заболевания, как анемия, тромбоцитопения и лейкопения. В организме наблюдается масштабное повреждение всех тканей организма, а также нарушение работы всех жизненноважных систем и органов. Заболевание как патологический процесс возникает на генетическом уровне.

Признаки синдрома
У синдрома Чедиаки-Хигаси ярко выраженные и очевидные признаки, которые сразу вызывают беспокойство за состояние здоровья ребенка. Первым внешним проявлением заболевания становится нарушение пигментации. Локализуется она в радужной оболочке глаза, на волосах, закрытых участках кожи, а также в области шеи. Помимо этого, наблюдается непроизвольное движение глазными яблоками, фотофобия, выражающаяся непереносимостью яркого света, нервный тик и блефароспазм.
На что стоит обратить внимание?
Бывают ситуации, когда все вышеперечисленные симптомы не вызывают тревоги у близких ребенка, однако следует учитывать, что пациент подвержен частым вирусным и бактериальным заражениям. Действие инфекций системное, то есть заражению подвергаются все системы и органы в организме. Довольно часто нарушения касаются мочевыводящей и пищеварительной систем, а также органов дыхания, слизистых оболочек и кожных покровов. Наиболее распространен патологический процесс грибкового происхождения, вызванный такими болезнетворными микроорганизмами, как стрептококк и стафилококк.
Повреждение кожи
У больных синдромом Чедиаки-Хигаси происходит обширное и повсеместное поражение кожи, которое характеризуется образованием папул и пустул, а также корочек патогенного характера, имеющих желтоватый цвет.
Подобные патологии кожного покрова нарушают температурный режим тела, а также сопровождаются отечностью и сильным зудом, образованием долго не заживающих язв. Поражение грибком проявляется в виде шелушения, налета на слизистой оболочке, гранулемами и свищами. Симптомы синдрома Чедиака-Хигаси неприятные.

Поражение органов ЖКТ
Поражение органов желудочно-кишечного тракта вызывает усиление диспепсии, что приводит к возникновению тошноты, рвоты, диареи и повышению температуры тела, которому сопутствует сильный озноб и лихорадка. В этой ситуации необходимо как можно быстрее восстановить водный баланс в организме. Иначе могут возникнуть непредсказуемые и опасные последствия для всего организма.
Поражение верхних дыхательных путей системного характера может сопровождаться ринитом аллергического генеза, трудноостановимым чиханием и приступообразным кашлем. Если не принимать необходимых мер, могут появиться влажные или сухие хрипы, которые четко слышатся при прослушивании. Также возможна лихорадка и резкое повышение температуры.
На мочеполовой системе синдром отражается в виде болезненного и трудного процесса мочеиспускания, скопления гнойных выделений, примесей крови в составе мочи, а в отдельных случаях сепсиса.
Если синдром Чедиака-Хигаси сопровождает анемия, она проявляется следующими симптомами: быстрая утомляемость и снижение работоспособности, бледность кожи и слизистых оболочек. Тромбоцитопения проявляется внутренними и внешними кровоизлияниями, а также нарушением функционирования все органов и систем.
Таким образом, можно сделать вывод, что синдром начинает атаковать самую ослабленную систему, постепенно разрушая ее, при этом присутствуют признаки основного заболевания.

Диагностика синдрома Чедиака-Хигаси
Чтобы установить наличие синдрома, необходимо провести тщательное и подробное обследование. Оно позволит не только идентифицировать саму болезнь, но и сопутствующие ему недуги. Поэтому на начальном этапе необходимо найти квалифицированного специалиста, который сможет дать правильную оценку всем имеющимся жалобам. Доктор изучает симптоматику и направляет пациента на тщательное обследование. Как правило, проводятся следующие лабораторные и клинические исследования:
- Общее, а также биохимическое исследование крови, которое позволяет выявить уровень содержания лейкоцитов и эритроцитов. На анемию укажет недостаток эритроцитов, в то время как сниженное количество лейкоцитов будет свидетельствовать о лейкопении.
- Специальное исследование крови на иммунодефицит также является частью мер для постановки диагноза. В данном случае проверяется соотношение Т- и В-лимфоцитов. Следует учесть, что при синдроме этот показатель может быть в норме.
- Ультразвуковое исследование. Достоверно укажет на наличие и преобладание в организме раковых клеток и патогенных новообразований. Кроме того, УЗИ позволяет выявить патологии сердца, брюшной полости и других важных органов.
- Рентгенологическое исследование грудины дает возможность проверить размеры миокарда и легочные структуры на наличие паренхим.
- Электрокардиограмма способна дать информацию о пороках сердца. Во время ЭКГ просматривается путь прохождения электроимпульса и его связь с областью миокарда.
После проведения полного обследования лечащий врач сможет сделать соответствующие выводы и правильно поставить диагноз, а также дать дальнейшие рекомендации пациенту.

Лечение
Синдром Чедиака-Хигаси является генетическим заболеванием, поэтому мер профилактики и предупреждения его развития в детском организме практически невозможно. Клинический исход полностью зависит от степени прогрессирования болезни на начальных этапах, при условии ранней диагностики его можно приостановить. По этой причине женщине необходимо ответственно подойти к вопросу планирования беременности. Если в роду имелись подобные заболевания, о них нужно сообщить лечащему врачу, а также сдать специальные анализы на выявление генетических заболеваний.
Что касается лечения патологии, то каких-то определенных схем не существует. Универсального лекарства, способного избавить от патологии навсегда, не существует. Поэтому терапия зависит от индивидуальных особенностей течения заболевания в каждом конкретном случае.

Профилактика
В основу терапии ложится тщательная профилактика вторичных инфекций. Это необходимо для предотвращения иммунных последствий для организма. Если ребенок уже подвергся заражению вирусами или вредоносными бактериями, назначается антибактериальная терапия. Также часто выписываются различные витаминные комплексы и иммуностимуляторы. К сожалению, синдром Чедиака-Хигаси чаще всего приводит к летальному исходу пациента до исполнения 10 лет.
Источник