
Синдром делеции 5 плеча короткой хромосомы

Данная группа заболеваний является результатом потери частей хромосом (делеций). Они могут стать причиной тяжелых врожденных аномалий, а также значительных умственных и физических отклонений. Подозрение на хромосомные синдромы делеции редко может быть пренатальным, однако они могут быть обнаружены случайно во время определения кариотипа по другим причинам. Послеродовой диагноз подозревается на основании клинических проявлений и подтверждается с помощью определения кариотипа, если делеция относительно большая, или с помощью других цитографических методов, таких как флуоресцентная гибридизация in situ или микроматричный анализ.
Синдромы хромосомной делеции, как правило, подразумевают значительные делеции, которые обычно заметны при определении кариотипа. Синдромы, включающие в себя незначительные делеции (и дополнения), которые оказывают влияние на один или более функционально сцепленных генов на хромосоме и незаметны при определении кариотипа, считаются синдромами микроделеций и дупликаций.
Заболевание обусловлено делецией короткого плеча 5 хромосомы (5p-минус, обычно имеющей отцовское происхождение); оно характеризуется пронзительным криком, очень похожим на крик котенка, который, как правило, слышен непосредственно в неонатальный период, длится несколько недель, а затем исчезает. Однако не у всех больных новорожденных наблюдается этот необычный крик. Больные новорожденные являются гипотониками и имеют малый вес при рождении, микроцефалию, круглое лицо с широко расставленными глазами, опущенные вниз косые глазные щели (с или без эпикантальной складки), косоглазие и широкий нос. Уши расположены низко, неправильной формы и часто имеют узкие наружные слуховые проходы, а также небольшие выросты, расположенные перед ушной раковиной. Часто возникают синдактилия, гипертелоризм и пороки сердца. Психическое и физическое развитие существенно замедленно. Многие больные дети доживают до совершеннолетия, однако имеют значительную недееспособность.
Делеция короткого плеча 4 хромосомы (4р) приводит к различной степени умственной отсталости; лица с большими делециями, как правило, сильнее страдают. Основные клинические проявления заболевания включают в себя умственную отсталость тяжелой степени, судорожный синдром, умеренную микроцефалию, антимонголоидный разрез глаз, широкий, клювовидный нос, птоз век, колобомы, расщелину твердого и мягкого неба, оттопыренные ушные раковины, гипоспадию, крипторхизм. Некоторые пациенты с синдромом Вольфа-Хоршхорна имеют также имунодефицит. Большинство больных детей умирают в младенчестве, а у тех, кто дожил до своего 20-летия, часто диагностируют тяжелую форму инвалидности.
Такие делеции могут быть заметны при определении кариотипа, однако, иногда, они бывают малыми и ультрамикроскопическими и могут возникнуть в любом теломере (конце хромосомы). Фенотипические нарушения могут быть слабовыраженными. Субтеломерные делеции могут быть ассоциированны с неспецифическим умственным нарушением и легкими дисморфическими чертами, а также у более тяжелобольных людей с врожденными пороками развития.
ПРИМЕЧАНИЕ:
Это — Профессиональная версия.
ПОЛЬЗОВАТЕЛИ:
Это — Пользовательская версия
© 2019 г. Мерк, Шарп энд Дом Корп., дочерняя компания «Мерк энд Ко. Инкорпорейтед», Кенилворт, Нью-Джерси, США)
Была ли страница полезной?
Источник
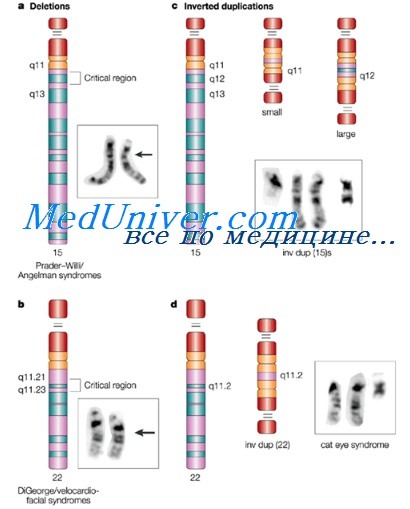
Структурные аномалии хромосом: несбалансированные перестройки, делеции, дупликацииСтруктурные перестройки происходят вследствие разрывов хромосом с последующим соединением в аномальной комбинации. Хотя перестройки могут происходить многими способами, все вместе они бывают реже, чем анеуплоидий; в целом структурные аномалии присутствуют примерно у 1 из 375 новорожденных. Хромосомные перестройки происходят спонтанно с низкой частотой, а также могут быть вызываны повреждающими агентами (кластогенами), например ионизирующим излучением, некоторыми вирусными инфекциями, многими химическими веществами. Подобно аномалиям числа хромосом, структурные перестройки могут присутствовать или во всех клетках человека, или в мозаичной форме. Структурные перестройки определяют как сбалансированные, если хромосомный набор имеет нормальный состав хромосомного материала, или несбалансированные, если отмечен дополнительный или отсутствующий материал. Некоторые перестройки стабильны и способны проходить через митотические и мейотические деления клетки неизменными, другие — неустойчивы. Чтобы быть стабильной, перестроенная хромосома должна иметь функциональную центромеру и две теломе-ры. Несбалансированные перестройки хромосомПри несбалансированных перестройках фенотип бывает аномальным из-за делеции или дупликации, а в некоторых случаях из-за их сочетания. Дупликация части хромосомы приводит к частичной трисомии; делеция — к частичной моносомии. Любое изменение, нарушающее нормальный баланс функциональных генов, может заканчиваться аномальными проявлениями. Большие делеции или дупликации, включающие несколько миллионов пар оснований, могут быть обнаружены при обычном хромосомном исследовании, в том числе при кариотипировании с высоким разрешением. Для обнаружения малых делеции или дупликаций обычно показаны более сложные анализы, включая FISH или микрочиповый анализ. Важный класс несбалансированных перестроек — субмикроскопические изменения области теломеры у пациентов с идиопатической умственной отсталостью. У нескольких процентов таких пациентов обнаружены небольшие делеции, дупликации и транслокации. Прицельный цитогенетический или геномный анализ теломерных и субтеломерных участков методами FISH или CGH-матриц может помочь в выяснении причин необъяснимой умственной задержки с целью генетического консультирования.
Делеции хромосомДелеция — потеря сегмента хромосомы, приводящая к хромосомному дисбалансу. Носитель хромосомной делеции (с одним нормальным гомологом и одним делетированным) моногамен по генетической информации в соответствующем сегменте нормального гомолога. Клинические последствия обычно отражают гаплонедостаточность (буквально — невозможность единственной копии генетического материала нормально выполнять функции, выполняемые двумя копиями) и зависят от размера удаленного сегмента, количества и функций содержащихся в нем генов. Цитогенетически видимые аутосомные делеции наблюдают с частотой приблизительно 1 на 7000 живых новорожденных. Малые, субмикроскопические делеции, обнаруживаемые микроматричным (биочиповым) анализом, более часты, но, как упоминалось раньше, клиническое значение большинства таких вариантов пока еще не полностью определено. Делеция может находиться в конце хромосомы (терминальная) или внутри хромосомного плеча (интерстициальная). Делеции могут быть следствием простого разрыва хромосомы с последующей утратой ацентрического сегмента. Кроме того, в некоторых случаях к делециям может приводить неравный кроссинговер между гомологичными хромосомами или хроматидами. Делеции также могут порождаться аномальным расхождением сбалансированной транслокации или инверсии, как показано ниже. При обследовании пациентов с дисморфиями и при пренатальной диагностике выявлено множество делеций. Знание функций генов, входящих в делетированные сегменты, и их влияние на фенотипические последствия существенно выросли в ходе реализации проекта «Геном человека». Методы окрашивания высокого разрешения и FISH могут выявлять малые делеции, невидимые на обычных метафазных пластинках. Если цитогенетическая окраска высокого разрешения позволяет идентифицировать делецию размерами не менее нескольких миллионов пар оснований, то делеций, не обнаруживаемые кариотипическим методом, и неопределенные делеций с фенотипическими последствиями стандартно обнаруживают методами FISH и биочиповым анализом с использованием зондов, специфичных для интересующей области. Дупликации хромосомДупликации, подобно делециям, могут происходить из-за неравного кроссинговера или ошибок расхождения хромосом в мейозе у носителей транслокаций или инверсий. В общих чертах дупликации оказываются менее вредными, чем делеций. Тем не менее, поскольку дупликация в гамете приводит к хромосомному дисбалансу (т.е. к частичной трисомии) и поскольку хромосомные разрывы могут нарушать гены, дупликации часто приводят к различным фенотипическим аномалиям. Хотя описано много дупликаций, достаточно изучены только некоторые из них. Тем не менее с дупликациями конкретных регионов хромосом связаны определенные фенотипы. Например, дупликация всего или части короткого плеча хромосомы 12р ведет к развитию синдрома Паллистера-Киллиана, при котором у пациентов отмечают характерные краниофациальные черты, умственную задержку и целый спектр других врожденных дефектов, вероятно, обусловленный трисомией или тетрасомиеи конкретного гена, расположенного в дуплицированной области. – Также рекомендуем “Маркерные и кольцевые хромосомы. Особенности” Оглавление темы “Аномалии хромосом”:
|
Источник
Синдром Клайнфелтера. Реципрокные транслокации и делеции
Синдром Клайнфелтера (47, XXY) встречается с частотой 1-2 на 1000 живых новорождённых мальчиков. Риск повторного рождения больного ребёнка небольшой.
Клинические проявления синдрома Клайнфелтера:
• Бесплодие — самое частое проявление.
• Гипогонадизм (недоразвитие яичек).
• Относительно нормальный пубертатный период (некоторым юношам помогает лечение тестостероном).
• Гинекомастия у взрослых мужчин.
• Высокий рост.
• Умственное развитие обычно в норме, но могут быть психологические трудности и проблемы обучения.
Реципрокные транслокации
Обмен материалом между двумя различными хромосомами называют реципрокной транслокацией. Когда этот обмен не сопровождается потерей или избыточным присоединением хромосомного материала, транслокация называется сбалансированной и фенотипически не проявляется. Сбалансированные реципрокные транслокации встречаются сравнительно часто — 1:500 в общей популяции.
Выявленная при обычном хромосомном анализе сбалансированная транслокация всё же может обусловливать потерю нескольких генов или разрыв одного гена, что приводит к изменению фенотипа, часто включающему умственную отсталость. Изучение повреждённых участков хромосом у таких пациентов является одним из путей выявления локализации специфических генов.
Несбалансированные реципрокные транслокации включают изменение объёма хромосомного материала и вызывают сочетание дизморфических черт, врождённых мальформаций, задержки развития и умственной отсталости. У новорождённых трудно сделать прогноз, но проявления обычно тяжёлые. Следует проанализировать родительские хромосомы для выяснения, является ли аномалия вновь образованной либо это следствие структуры родительских хромосом.
Обнаружение сбалансированной транслокации у одного родителя указывает на риск повторного рождения больного ребёнка, существует возможность антенатальной диагностики с помощью забора ворсин хориона, или амниоцентеза, а также тестирования родственников.
Делеции
Делеции — другой тип структурного нарушения. Потеря части хромосомы обычно приводит к физическим аномалиям и умственной отсталости. Деления может представлять собой потерю концевой или, реже, внутренней части хромосомы.
Примером делеции является потеря концевой части короткого плеча хромосомы 5, называемая 5р-, или моносомия 5р. Поскольку у больных новорождённых детей плач похож на звук кошачьего крика, этот синдром также называется «синдромом кошачьего крика». Следует исследовать хромосомы родителей для выявления носителя сбалансированной транслокации хромосомы.
Известно, что все большее число синдромов обусловлено делециями хромосом, которые слишком малы для выявления в обычном цитогенетическом анализе. Такие субмикроскопические делеции можно выявить с помощью FISH, используя ДНК-пробы, специфические для определённых хромосомных участков. Синдром Ди Джорджи развивается при делеции хромосомы 22 на участке 22qll. Синдром Вильямса — ещё один пример микроделеции с потерей хромосомного материала на длинном плече хромосомы 7 на участке 7qll.
– Также рекомендуем “Аутосомно-доминантное наследование: принципы, примеры”
Оглавление темы “Генетические болезни детей”:
- Синдром Тернера: причины, клиника
- Синдром Клайнфелтера. Реципрокные транслокации и делеции
- Аутосомно-доминантное наследование: принципы, примеры
- Аутосомно-рецессивное наследование. Близкородственный брак
- Х-сцепленное наследование: принципы, примеры
- Синдром ломкой Х-хромосомы: принципы, примеры
- Митохондриальное и цитоплазматическое наследование. Импринтинг и дисомия от одного родителя
- Полигенное или мультифакторное наследование: принципы, примеры
- Анализ ДНК детей. Методы
- Пороки развития детей: патогенез, классификация
Источник
Хромосомные аберрации, или аномалии в структуре или количестве хромосом, являются одними из наиболее распространенных, наиболее понятных и относительно простых для исследования генетических причин бесплодия.
Статистика по хромосомным аберрациям
- Частота хромосомных аберраций у мужчин с низкой фертильностью, обнаруженных в соматических клетках (обычно проверяются мононуклеарные лейкоциты крови) в общей популяции новорожденных детей, составляет примерно 0,7%.
- Среди фенотипически нормальных мужчин с нормозооспермией, но с репродуктивной недостаточностью, частота значительно выше и составляет около 3%. Чаще всего это аберрации, касающиеся структуры аутосомных хромосом (частота взаимных транслокаций составляет 0,93%, робертсоновских транслокаций – 0,46%, инверсий – 0,23%).
- Частота аберраций Х- или Y-хромосомы у этой группы пациентов оценивается в 1,4%, наиболее частые случаи – клеточный мозаицизм числа хромосом (0,93%).
- У фенотипически нормальных мужчин с азооспермией (необструктивной) хромосомные аберрации в лимфоцитах выявляются примерно в 13,2% (в 20 раз чаще, чем в общей популяции новорожденных детей).
Среди хромосомных аберраций мы выделяем числовые аномалии, например, изменения числа половых хромосом: синдром Тернера X0, синдром Клайнфельтера XXY, синдром XXX, синдром XYY, и структурные аномалии – делеция хромосом, хромосомный дефицит, инверсия, дупликация, транслокация, разрыв центромеры. Хромосомные транслокации могут привести к снижению фертильности, самопроизвольным выкидышам и врожденным дефектам. У субфертильных мужчин явно повышенный риск переноса хромосомных аберраций, и их обнаружение важно для оценки бесплодия.
Наиболее распространенный тип аномалии кариотипа у субфертильных мужчин – синдром Клайнфельтера (СК), который вызван наличием дополнительной Х-хромосомы Реже хромосомные аберрации выражаются в виде других числовых (две, три, четыре дополнительные хромосомы) или структурных изменениях. Таким образом, KS проявляется в кариотипе 47, XXY или других кариотипах, например 48, XXXY, 48, XXYY, 49, XXXXY, 46, XY / 47, XXY, 46, XY. Дополнительная Х-хромосома может быть материнской или отцовской из-за нерасхождения во время мейоза.
Кариотип с синдромом Клайнфельтера
Среди всех мужчин с азооспермией примерно 14% пациентов с синдромом Клайнфельтера характеризуются маленькими яичками и высоким уровнем гонадотропинов. Азооспермия при синдроме Клайнфельтера поражает подавляющее большинство пациентов (почти 98%),
Другая числовая аномалия половых хромосом у женщин – синдром Тернера, который более чем у 80% пациенток проявляется гонадным дисгенетизмом – первичной недостаточностью яичников (сексуальный инфантилизм). Синдром Тернера, помимо ряда других симптомов, характеризуется:
- задержкой полового созревания;
- отсутствием телархе и лобка;
- первичным бесплодием;
- пониженным уровнем эстрогена;
- высокой концентрацией гонадотропинов ЛГ и ФСГ.
Следует подчеркнуть, что примерно у 10-20% пациенток функция яичников частично сохраняется: менархе присутствует, но может исчезнуть. Женщины с сохраненной функцией яичников, вторичными половыми признаками без аномалий и фертильные женщины встречаются редко. Современное развитие генетических методов позволяет обнаруживать синдром Тернера во внутриутробном периоде до 30% случаев.
Кариотип 47, XYY (синдром Якобса), ранее называвшийся синдромом супер-мужчины, является второй полной анеуплоидией половых хромосом, вызванной трисомией половых хромосом с дополнительной Y-хромосомой. Риск анеуплоидии у мужчин с 47 XYY не подтвержден.
Другое заболевание – синдром кариотипа 46, XX, наблюдаемое в основном у мужчин с азооспермией с частотой 0,9%. Фенотип аналогичен синдрому Клайнфельтера. Следует подчеркнуть, что мужчины с синдромом Клайнфельтера обычно не являются умственно отсталыми и всегда бесплодны.
Около 0,9% мужчин с азооспермией (и нормальным фенотипом) имеют кариотип 46, XX (SRY +), который является результатом транслокации между Xp и Yp и включает ген SRY, важный для определения мужского пола. Вторая категория, очень редкая, – это SRY – у мужчин с нормальным фенотипом и с бесплодием, или в случаях истинного гермафродитизма у людей неясного пола. В качестве причин указываются мутации генов, расположенных за пределами Y-хромосомы, участвующих в каскаде, определяющем пол.
Большинство исследований по анализу хромосом в сперме касается лиц мужского пола с аберрацией в структуре соматических хромосом. У мужчин они не имеют фенотипических эффектов и видны во время гаметогенеза. Это результат мультивалентности. Среди расщепления мейотических хромосом в сперматозоидах у носителей транслокаций можно выделить взаимные и робертсоновские транслокации.
Цитогенетические тесты, проводимые у женщин в рамках рутинного скрининга перед вспомогательными репродуктивными технологиями, показывают некоторые аномалии, такие как реципрокные аутосомные транслокации, о которых говорят, когда два фрагмента двух разных хромосом отламываются и меняются местами. Частоты реципрокных транслокаций как у женщин, так и у мужчин существенно не различаются. Например, этот тип аберрации встречается примерно у 1,0% женщин перед ЭКО, а перед ИКСИ – у 0,7%. Большинство реципрокных хромосомных транслокаций наследуются от родителя.
У большинства мужчин, гетерозиготных носителей сбалансированной реципрокной хромосомной транслокации, гистологическая структура, а также гаметогенная активность и количественный состав сперматозоидов являются нормальными, а транслокации, вызывающие полное ингибирование сперматогенеза, встречаются редко. Сильное влияние на сперматогенез оказывает переносчик транслокаций, которые характеризуются расположением хромосомного перелома в непосредственной близости от центромеры, а также транслокаций с участием акроцентрической хромосомы – в этом случае, как было обнаружено в многочисленных исследованиях, наблюдается уменьшение количества сперматозоидов в эякуляте, олигозооспермия или их полное отсутствие. Кроме того, носители транслокации между Х-хромосомой и аутосомой также являются субфертильными,
Робертсоновские транслокации (центрическое слияние) являются наиболее распространенными структурными транслокациями у людей и влияют на фертильность, особенно у мужчин.
Робертсоновские транслокации хромосом
Робертсоновские транслокации обнаруживаются у небольшого (0,2%) процента обследованных женщин с проблемами фертильности и не играют существенной роли в патогенезе женского бесплодия. В популяции субфертильных мужчин 0,8% являются носителями центрического слияния, что в 9 раз больше, чем у фертильных мужчин.
Робертсоновская транслокация затрагивает только акроцентрические хромосомы (13-15, 21 и 22), где перелом происходит на центромере или рядом с ней. Соединение двух акроцентрических хромосом дает одну, часто с двумя центромерами, а короткие плечи хромосом, участвующие в слиянии, чаще всего устраняются. Транслокации хромосом 13 и 14, а также 14 и 21 являются наиболее распространенными транслокациями у человека (частота транслокаций Робертсона составляет 1/1500 человек). Они могут передаваться от отца или матери.
Перенос Робертсоновской транслокации не влияет на фенотип, но оказывает сильное влияние на фертильность из-за возможности нарушений гаметогенеза и / или из-за образования несбалансированных гамет. Проблемы с фертильностью у мужчин – носителей этой транслокации, возникают в результате нарушений (разной степени) сперматогенеза, непосредственно связанных с аномалиями в процессе мейоза. Это приводит к образованию значительного процента несбалансированных гамет, которые участвуют в оплодотворении, вызывая аномальный кариотип полученных эмбрионов.
Делеции Y-хромосомы
Среди делеций Y-хромосомы, которые представляют собой вторую группу генетических нарушений, влияющих на фертильность, мы выделяем микроделеции Y-хромосомы и делеции AZF.
Делеции AZF вызывают нарушения сперматогенеза в виде азооспермии и тяжелой олигозооспермии. Микроделеции длинного плеча Y-хромосомы нарушают процесс сперматогенеза примерно у 5-15% мужчин с азооспермией или тяжелой олигоспермией. Эти расстройства передаются по наследству от отца к сыну. Эти микроделеции мешают процессу сперматогенеза, поскольку гены, кодирующие белки, участвующие в производстве сперматозоидов, расположены в области AZF.
Структура AZF-локуса Y-хромосомы
Следует подчеркнуть, что существует пропорциональная зависимость между степенью делеции и степенью нарушения выработки спермы – сильные делеции связаны со значительным нарушением сперматогенеза. В 80% делеций AZF происходит удаление AZFc. Что касается других делеций этого типа, то:
- делеция AZFa встречается в 0,5-4% случаев;
- AZFb – в 1-5%;
- AZFbc – в 1-3% случаев.
Комбинация AZFabc сосуществует с другими расстройствами, такими как синдром 46, мужской XX или Iso (Y). Носительство вышеуказанных делеций связано с различными фенотипами, включая азооспермию, хотя есть многочисленные случаи отсутствия отклонений в параметрах спермы. Примерно 2/3 мужчин с делецией фрагмента AZFc демонстрируют наличие сперматозоидов в сперме. В случае делеции AZFc, проявляющейся азооспермией, активность семенного эпителия яичка, формирующего сперматозоиды, сохраняется, и результаты TESE (извлечение спермы из яичек) положительные.
Напротив, в случае делеции фрагмента AZFb шансы получить сперму методом TESE невелики, и у мужчин с делецией AZFa при диагностической биопсии часто обнаруживаются только клетки Сертоли. Более того, мужчины с делецией AZFc, gr51 / gr51 подвержены повышенному риску зачать ребенка с синдромом Тернера (45, X) и подвержены риску развития первичного рака яичка.
Продолжение статьи
- Часть 1. Генетические причины бесплодия – статистика, цитогенетические исследования.
- Часть 2. Хромосомные аберрации, делеции Y-хромосомы при бесплодии.
- Часть 3. Генные мутации, полиморфизм генов при бесплодии.
Поделиться ссылкой:
Источник