
Синдром делеции по длинному плечу 18 хромосомы

Синдром делеции 18-й хромосомы. Синдром кошачьих глаз – синдром Шмида—ФраккароСимптомокомплекс, обусловленный нехваткой генов, локализованных в длинном плече 18-й хромосомы, описан в 1964 г. I. De Grouchy и др. Частота в популяции не установлена. Синдром включает задержку психического развития, микроцефалию, низкорослость, снижение слуха, деформацию ушных раковин и наружных слуховых проходов, микрофтальмию, гипертелоризм, колобомы радужной оболочки, нистагм, ретракцию средней части лица, плоское переносье, «карпий рот», высокое или расщепленное нёбо, аномалии стопы и бедер, клинодактилию, пороки сердца, почек, половых органов. Синдром верифицируется кариологическим исследованием, при котором обнаруживают простую или замаскированную фрагментом делецию.
Синдром кольцевой 18-й хромосомыСимптомокомилекс, обусловленный частичной нехваткой генов, локализованных в теломерных участках 18-й хромосомы, описан в 1964 г. I. De Grouchy и др. Частота в популяции не установлена, однако известно, что она не зависит от возраста матерей. Дети рождаются с низкой массой, в дальнейшем резко отстают в физическом и психомоторном развитии. Характерна микроцефалия, описывают гипертелоризм, уплощенный затылок, эпикант, деформацию ушей и глухоту, косолапость, синдактилию, аномалии бедер. Синдром «кошачьих глаз» (синдром Шмида—Фраккаро)Симптомокомилекс обусловлен избытком гепов, локализованных, повидимому, в 22-й хромосоме. Частота его в популяции не установлена. При кариологическом исследовании, по крайней мере в части клеток, обнаруживают маленькую добавочную хромосому. – Также рекомендуем “Синдром делеции хромосом группы G. Синдромы дупликации-делеции с частичной трисомией 9-й хромосомы” Оглавление темы “Наследственные синдромы в неврологии”: |
Источник
Ö11(à
k:`èË^º}m_5(Q`JIÑ;ȲÚY
4ïùc.*ZSfà|ÌÖxxûd
à´Ø¯¶¢q%#
¤B½íôþ,ºÞÖ»’²%å£ÕÑ
î³*U¦0dÿÛL2Ó:
¦@K¯*6[
2KVǤ:Ö6Vx¬-UänRêVj®-¸(û7I°ÏóÖÑ’d0øtÆú¦*.îÂB l4¢µ{Þ¹ØÇS°Ñëu+ïmç¾³â³Ù`Ô ¾
묲P³ÙEúºìê²Q$ÎAöªÖGêõ0mz$nVJf8«¨”l3ýXA½D0Nù,KÆG0íñ ?xí;ȬÒJ£ !+f,óë5v Y |’CGÆpP m»ìÕ°X
) bôÞç½h0 ¡+: lÕ2V|
L,`Í hѤg̶¹PÛÆ6 ÷jk·YãÕ3ËÔ*
ò4¶u2Ökt
sÂêX&álYgGãØâ`´â ñÉ,¦·ò[>
½æâëòØdÉÕº²=O]î§É^Ðì§Y&qC [ç”1ôÂ`¦¼SÄÔ$¨K>]»äs ½p*ÿ䪴SÏ$’ÌìÏÑÌÔCÄ¢Û1,PÐ`?^CË0È%õ9§ÜßÉi´|kÊÜ~»hz¡cí+äS!×gË÷Fw«ú¾¼ùÒ¼ô û?£¤Aý?RjFÖ¾ëñ¬¤%ºêæìÈK8+aäóì)cÙs±Ø¦){!«àøTÏ ‘|Qi¤µüé3NÓíòþÄÚ±q´Cú槒o×N@nkw>édáZÌê_UEK4¬ê[-l¡U1SÑ,N¬ìRô°^¤«5kÏçh$
aó~s/Ç>JÆp~
’û÷(i1(>v:Jͤm®Epû¤@èÖãäí³wPðu«LÉ¡]±9½Èà[ÑéÖO3çÊaJ·3hmd®?
É 3kYxßÖÆi ÞÓ[á¸øu`h
ý³þç³ðï±[]·ÑmNpæÀ}_¸ZÁÊ+L¶Ø§8A|UO³·9k7Y ×Dv°Âfw ùÍ«lï=!ÈÞÌpñaÎjÙ_öº·ëîÊ$3_°F#ЦÆlËU Z±ë?ý’Ji”¤ &çFá*r´K>çüÝdn6~^ߤ § }¿ ³vÀÏ`§M#+È,Òi[àFõüPÓ÷ѽ±©ãßÎ4^v
NNQ¿bÃq(áW£Vó5 {ßÒV
+(t³
>Äô%jáPÏO-;°9»}YYlµý¼Ï ¾ü¬HqÄN=é;#GEç©æ]?è(VÈ^j£I
Ïu}Ó9I#£cJ~»&úeM 0n6»½]ÄÌ1¬FlÀÃYÊýøÀoI ÌkôpZªÞ’’ïÄÊXÅ?EHuUTIß.kuQ0p̧LôʪeÂ;=ï~P¶!n5pYtò!ÒÜ0ïÕAéIhýr±ßÔç’çÜã¼½ýúòe©VxÃÈóí,hb§oæsFøÎùóÛÕÏõ &x`Zk?;2«ÀTfxÍ 0¯°«õI£Ü/®qtFBÀºÖ¿M¯2-êp:jPf-LGùì[Bï>ÈRç(Æñg’ÐèM0å$ê3iµÏ¼ËçÈf4^îèÙ$V,eîX’àp¶?_¾º§|’ÃJ×áóÉ(EÍc ý³ó~Æå¸îcÙÞeö,ì’:öâÆÚ°9öÉ} rðÑå+ù>8i~hÊÐX ¤.àÁC’;õ³ãÒ£Fo®ºaôìfCòuíW)¥TMN$æT_Ý1í²{)ré¹N”ósv+Pµ+
FÃæ~ÕÁ`b®z Å.ÙÜ*¡Ú2a@@Æìc.5sè¼ ;Û[y=>¹ræÓZÖîÊ
3ëT!ZB¿Ö`ø9sB{§×|’·Nª´Ws¬¦iâêhÜz,5rö&m ÖeÓâ/BñUà
Rm½!Ø_
÷ÀÝWYÙ
ã×ïË(sÓÕöäå§çb,H{×4F -w*ÉFÑà^Rg©¼é³{ha¶ÏîùzÆé¾ý¤ãÇ¿b éb³·PS_N+>´%³”²_Y þÞió©à¡ªF}õôëÖƳ.øº
ÐÀÿÜ æH¹[hu$ûéèH/Ë>
$æ2;½·ÄÀÉü(²ñ«-IäØNËIGoÓ~Jv£ÅKÕâÕ:-ûG2äq¹Ù99K(É`ѬÕ66khx¥¨3$ÐW]z¼ÁæE½«e_ºãLIh>.ÖDM¼¸t¬zºG+¸{
O&µ²ÔÊU»fÙHV6(7g÷gjÄb®2¢Iñ%*e/§ÖóÕ?˵+ïþ]Ä|({¤ÐÿÕà ¿4t sýFø4ÀXË:IÔ Ñ Ö;¡þBÛ
oòÉ_$íÇø”¹uÚXU_¿”Åá5h lêÎ%f«kÙ(?½ìΔÅp¤Y2_HÜ*ÑVá;»ùAêî¤å42q”622ÏÈR5(ÕM×wÿ冀 ÝECé$WÔZëJC{CvwC`WÈ÷OAG
iÝ0¦K½úÖÄÁj×k¥`ãG[ëÒHPîƬ1õHEk4Î=x3ÿ4(Ì~s½ÕæÈÔd2ÊzÕ`0f²Ù)¾&.¢ãj÷Ó:ªô©4z§ElJÏûÑsI«¡n÷N, uoðÓ^î©?A¥`(ö£[ ŵöÃ
^¨t~}YÓt¶06×gbpmZ^hZttÇxå@¿bè±ÒÂ(÷©êXßÉÒÙ}«Ç®Æ¢Ú%+ñβûu/øFì|÷®PTHg1>ÀTÍ- ÝˤåëM ëÎE:cõtÆEö:XÿH6â¿]s8IÍÛÙ¤®ëSÀÎîÿlL,»wwMⶫު syáïÝ*-V®å ¹TRÓsÛèhw«
ĨJ¡ìS23=_6ç8HÆÌ Ê~Cq÷ It»Æ²3âÓªÓÀ¢=ÝeÎÛ°N= W°Êä®
yÃUo¹Êw-{îi+F¿_m»M
Ò@Z
zÊíh>ÊdÕð-yC3%³ÂÓ¥½÷ï§ÁeYÑQS¸Q+»êÏ
·µêmkøÓÙûÊ”Ðo*9ÁL«2ÇY|ï5¾6&з¤û¨ßÄ!XfEéZò4
úë=mzªMìv>ܵÍ_GçÍ¢nvwû«#ºB¬.²f¹Ô_bÝõ³÷9E÷4¦¨6Û#
ȡ׫}l/¿æ(tþIµ£f³8HCVaMÈáÉg9¦¹°;Üj¢H;Í^¿$ÝX6ëfån¦YÄzv%«Î?bâ¨aÙUÁäbè7ÚHãã¥@°Fæ]6ÛëîLÎÂ0Ð]`¨`sÄFI·Ïæw´¥¥ sYÅÑd9Ï«W4NyÔL8`UU18k+W0dÊ_ÑÀ£D¯ÓÓRô¹Z[¨1J,úhpK!>
¹V¥;èÐ}¿7_døþ:=m Õx£ÕÈEno¯f½ÏS|¦VF6
⬻õ´s5pÊ¢ÙÎs,åÙiJ.êÂÎ4Wc,·Ë!m.7AîÚ%U×L¡¤ï;>¦©±0ïA*’Ôb[ZCRã³ö_WF-
î1yê
/ÓM7£×ËDM¾:[pÞÉgwCh±yÀ@ÈY)Ê·~ ¯¡ÌQâòèJ,ÕSæ[³ êÚÕaÈÚt|´ o£²[‘
#(¶É¨&_ÛYO¬]mk¼.9BÊ´ìÀ¯¸Ü1åQá¡PFæ Ê´í²+üª#8« Ë ²OÃTåîúÂ;lçá`ʨÜ!m{%n²Uÿêû¦ðzÌ*|ZDBÊmPÒÍ1ÅñÊ©7″Ù”°Y
Íõ§k|b¾ryä½z
Ûâv&ú)8lýxÉæeR¶3ÿ¨OîdþÊBçù?N!3f°7`8lµ»0¦á AÑßöÚ¡¡Ã¤@±FYE}@^[Õé
1 +` ÀÒûR´d8ý)¥woH©®Ínß åu#]è(;PZèvY*ëJÉÊ3õÏ0[9WeÍçb2_ÆæDKÍÂÊÉæÂú.k~ÞÃi*öA,¨# áþRËK¨ÉþYH2±ëû>¹¢¯Äá%¥ÞÎö ÌÕ^Úaqà (
+ûXCöåÁÎ)`;Ì Ö^õ&îbòºàÈߣ¹üŬO5ÆK(íê¼q˽ØhUIê)0!ç}ü)v7Oî3¤´ÓÅγ{æt)`ÁxI¦X·ªINÞNð.rUl½fLÍ}¥ß”ë¼ZdVÚf| dgá/ºµ²Õ·f篲Á*ųµG©¿J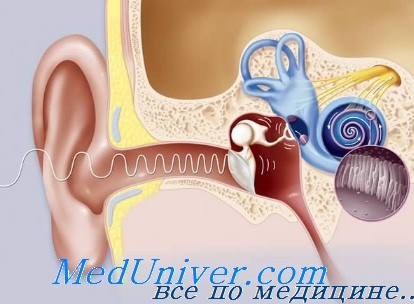
Серповидноклеточная болезнь с глухотой
Серповидноклеточная болезнь является относительно частой гемоглобинопатией у негров. Установлено, что около 7—9% американских негров гетерозиготы по гемоглобину S. Таким образом, гомозиготы встречаются с частотой 1:400.
У гомозигот вначале может появиться только анемия с небольшой спленомегалией или даже с нормальной селезенкой. Но у них наблюдаются экстрамсдуллярное кроветворение и гиперплазия костного мозга. У больных могут возникнуть повторные приступы слабости, утомляемости, болей в животе, отсутствия аппетита с желтухой и бледностью. С возрастом у них могут развиться расширение сердца, изъязвления ног, асептический некроз бедренной кости и гематурия.
Morgenstein и Маnасе описали у больных с серповидноклеточной анемией двусторонний умеренный дефект слуха, выраженный резче на высоких частотах. Исследования височной кости показали дегенеративные изменения в кортиевом органе и сосудистой полоске, совместимые с ишемией. Костный мозг внутри слуховых косточек был гиперплазированным и эрозивным. Todd с соавт. обнаружили у 22% больных с серповидноклеточной болезнью на Ямайке, по сравнению с 4% в контроле, нейросенсорный дефект слуха, достигающий по крайней мерс уровня 25 дБ.
Мужчины и женщины были поражены одинаково часто. Нарушения слуха выявлялись постепенно, первично нарушалось восприятие высоких тонов. Авторы считают, что дефект слуха развился в результате тромбоэмболического процесса. Serjeant с соавт. исключили сужение внутреннего слухового прохода как этиологический фактор потери слуха при серповидноклеточной болезни.
– Также рекомендуем “Актуальность изучения зрительного восприятия устной речи. Чтение с губ”
Оглавление темы “Генетические болезни с глухотой”:
- Синдром Жервелла и Ланге-Нильсена. Кардиоаудиторный или сурдокардиальный синдром
- Ушная-зубная или отодентальная дисплазия. Липодистрофия лица, плеч и кисты костей с глухотой
- Семейная отогенная интоксикация стрептомицином. Синдром Тернера с глухотой
- ХХ-дисгенезия гонад с врожденной глухотой. Трисомия 21 и 13 с глухотой
- Трисомия 18 и делеция длинного плеча хромосомы 18 с глухотой. Серповидноклеточная болезнь с глухотой
- Актуальность изучения зрительного восприятия устной речи. Чтение с губ
- Возможности развития навыка чтения с губ у глухого. Изучение процесса чтения с губ
- Видимые признаки артикуляции русских фонем. Группы артикуляции букв
- Особенности артикуляции согласных. Артикуляция гласных букв
- Чтение с губ неударных гласных букв. Артикуляция губ при произношении
Источник
Хромосома 18 представляет уникальную модель для изучения влияния на фенотип как избытка ее генов (синдром Эдвардса), так и их недостатка. Поскольку полная моносомия по хромосоме 18 не описана, речь может идти лишь о частичных моносомиях. Утрата материала хромосомы 18 может проявляться как делецией короткого плеча (18р-), так и делецией длинного плеча (18q-), Существует и третья форма моносомии -кольцевая хромосома 18 (18г), при образовании которой теряется часть материала обоих плеч, однако особенности поведения кольцевой хромосомы в митозе могут вести и к существованию линий, частично трисомных по некоторым локусам хромосомы 18. Практически, однако, при 18г отмечаются лишь признаки 18р- и I8q- Судя по количеству публикаций, создается впечатление, что каждая из форм частичной моносомии 18 встречается примерно с равной частотой. Однако если учесть, что 18q- проявляется более четким клиническим синдромом, чем 18р-, а следовательно, легче диагностируется, то следует считать, что 18р- более частая форма аберрации, чем 18q-.
Хотя клинически в рамках частичных моносомии 18 удается выделить 2 синдрома, 18р- и 18q-, цитогенетичсские характеристики обоих имеют много общего, что позволяет рассматривать их вместе.
В большинстве случаев делеция хромосомы 18 является результатом хромосомной мутации в гаметогенезе одного из родителей, поскольку кариотипы родителей чаще всего оказываются нормальными. В ряде наблюдений имеет место мутация в соматических клетках, о чем свидетельствуют обнаружение у ребенка мозаицизма с наличием и нормального, и аберрантного клона. Б сравнительно редких случаях утрата материала хромосомы 18 (чаще утрата коротких плеч) происходит в ре зультате спорадических транслокаций. Значительно чаще причиной возникновения аберрации у ребенка являются сбалансированные реципрокные транслокации у одного из родителей. Наконец, что более редко, описаны утраты материала хромосомы 18 в результате наличия у одного из родителей инверсий или вставочных транслокаций. Таким образом, цитогенетика частичных моносомии 18 достаточно гетерогенна. Вместе с тем в любом варианте материал коротких плеч всегда утрачивается почти полностью, тогда как при делеции длинного плеча наряду с терминальными делениями возможны и интерстициальные.
Дети с обеими формами моносомии рождаются обычно в срок (средняя продолжительность беременности не отличается от нормы), в то же время масса таких детей обычно несколько ниже нормы. По нашим данным, и при делеции 18р- , и при делеции 18q- средняя масса детей при рождении около 2800 г.
В среднем возраст родителей при рождении ребенка со спорадической делецией 18q- не отличается от возраста родителей в популяции, тогда как у родителей при делеции короткого плеча хромосомы 18 он значительно повышен. Он оставляет 31,2 года для матерей и 35,3 года для отцов, что почти на 5 лет выше среднего возраста при деторождении и примерно соответствует возрасту родителей детей с синдромом Эдвардса. Этот феномен следует объяснить скорее всего тем, что при делеции 18р – «поломки» происходят чаще всего в непосредственной близости от центромеры.
Прн всех формах частичной моносомии 18 девочки поражаются чаще, чем мальчики, примерно в соотношении 1,6:1.
В этой связи уместно отметить, что при трисомии 18 девочки поражаются также значительно чаще. Очевидно, между хромосомами X и 18 существует какая-то связь, н нарушение функций хромосомы 18 балансируется воздействием Х-хромосом.
Фенотипические изменения при кольцевых хромосомах 18 складываются за счет утраты части материала и коротких и длинных плеч, поэтому здесь наблюдается сочетание признаков как 18р-, так и 18q-, и по всем клиническим параметрам пациенты с кольцевой хромосомой 18 занимают промежуточное положение между больными с делениями короткого и длинного плеча.
Источник
Синдром трисомии 18-й хромосомы. Синдром Эдвардса в неврологииСимптомокомплекс, обусловленный избытком генов, локализованных в 18-й хромосоме, описан J. Edwards с соавторами в 1960 г. Частота синдрома составляет 1 : 3000 — 1 : 4500 живорождений. Более 70% всех детей с трисомией Е — девочки. Это, вероятно, связано с лучшей их выживаемостью. Девочки живут в среднем 282 дня после рождения, в то время как мальчики — только 58,5 дня. Беременность нередко протекает с токсикозом, многоводием. Дети рождаются в асфиксии, несмотря на нормальное течение родового акта. Плацента уменьшена в размерах, с атрофичными дольками и единственной пупочной артерией. Клиническая картина характеризуется большим полиморфизмом симптомов, хотя ни один из них по существу не является специфичным для трисомии Е. Каждая из аномалий может встречаться и при других хромосомных аберрациях, однако их сочетание придает больному довольно характерный внешний вид, что позволяет диагностировать трисомию Е уже в периоде новорожденности еще до цитологического обследования.
Дети рождаются очень слабыми, с низкой массой, плохо сосут, не прибавляют в массе. Череп деформирован, долихоцефалической формы, небольших размеров, затылок выступающий, прямая линия скошенного лба и носа, гипертелоризм, эпикант, микрофтальмия. Уши низко посажены, асимметричны, недоразвитие завитка и противозавитка ушной раковины, микрогнатия, гипоплазия нижней челюсти, готическое нёбо, крыловидные складки на коже шеи. Типичными являются флексорные контрактуры II и V пальцев кисти: средний и безымянный пальцы согнуты и приведены к ладони, мизинец и указательный накладываются на них сверху. Грудина укорочена, таз узкий, вальгусные стопы, гипоплазия отдельных групп мышц, врожденные вывихи тазобедренных суставов, крипторхизм. Мышечный тонус снижен, безусловные рефлексы угнетены, быстро истощаются. После периода новорожденности прогредиентность в физическом и психическом развитии больных отсутствует. При патоморфологическом исследовании обнаруживают дефект межжелудочковой перегородки, открытый артериальный проток, диафрагмальную грыжу, меккелев дивертикул, стеноз привратника, гидронефроз, дольчатую почку, агенезию почек. В головном мозге находят микрогирию, олигогирию, недоразвитие мозолистого тела, атрофию нервных клеток мозжечка и красного ядра. Характерны дерматоглифические изменения: поперечная складка на ладони, увеличение числа дуг на пальцах, отсутствие пальцевых трирадиусов с, b и d, слабо выраженный узор на мизинце, продольная складка стопы. Окончательный диагноз устанавливается после цитологического исследования, когда выявляется наличие целой дополнительной 18-й хромосомы или ее фрагмента на одной из аутосом. – Также рекомендуем “Синдром Патау в неврологии. Синдром трисомии D2″ Оглавление темы “Наследственные синдромы в неврологии”: |

Источник