
Синдром гипофизарной карликовости с нейросенсорной глухотой

Гипоталамо-гипофизарная карликовость с глухотой. Мукополисахаридозы: синдром ГурлерО 2 сестрах с гипоталамо-гипофизарной карликовостью и нейросенсорной глухотой сообщили Winkelmann с сотр. Отставание в росте впервые было замечено при поступлении в школу. У взрослых сестер рост был равен 139 и 146 см. Эндокринная система. Ни одна из девочек не достигла половой зрелости, ни у той, пи у другой не появились волосы на лобке и не развились молочные железы. У обеих сестер отмечались первичная аменорея и инфантилизм наружных и внутренних гениталий. Вестибулярная система. Результаты исследований не опубликованы. Радиоиммунологическое исследование уровня гормона роста в плазме крови показало резкое его снижение (3,0 нг/мл против 30 нг/мл в норме). При стимуляции инсулином уровень гормона роста повышался, но не выше чем до 5,0 нг/мл. Содержание фолликулин-стимулирующего (FSH) и лютеинизируюшего (LH) гормонов соответствовало препубертатному уровню. Усвоение радиоактивного йода, а также выделение кортизола и кортикостерона были нормальными. Уровень 17-гидрокси- и 11-гидрокси-кортикостероидов в моче был нормальным, даже после стимуляции АКТГ.
Мукополисахаридозы: синдромы Гурлер и ШейеМукополисахаридозы — наследственные болезни мукополисахаридного обмена. Дефект активности различных генетически контролируемых путей лизосомалыюй деградации ведет к внутриклеточному накоплению недеградированных кислых мукополисахаридов и к относительно схожим клиническим и скелетным изменениям. Фенотип наиболее резко выражен при синдромах Гурлер и Марото— Лами и менее резко при других мукополисахаридозах (McKusick, Spranger). В связи с малым объемом этого текста мы сможем только кратко в общих чертах представить клинические и лабораторные данные о тех мукополисахаридозах, которые сопровождаются глухотой. При синдроме Шейе (МПС-1-Ш), аллельной форме синдрома Гурлер, черты лица несколько грубоваты, наблюдаются нижнечелюстной прогнатизм и опущенные углы рта. Нос широкий. Начинающееся в молодости прогрессирующее помутнение роговицы приводит обычно к снижению остроты зрения в четвертом десятилетии жизни. Больные слегка отстают по росту от сверстников. Интеллект нормальный. Кисти и стопы широкие, пальцы па руках и ногах фиксированы в «когтеподобном» положении. Подвижность всех суставов ограничена. Часто наблюдается синдром «туннеля запястья». У большинства больных отмечается регургитация аорты. – Также рекомендуем “Синдромы Хантер, Санфилиппо, Моркио, Марото — Лами. Орган слуха при мукополисахаридозах” Оглавление темы “Наследственные болезни с глухотой”:
|
Источник
Болезнь Тея — Сакса с глухотой. Молочная ацидемия, миопатия и низкий рост с глухотойМы полагали, что твердые данные о нарушениях слуха при разных мукополисахаридозах малочисленны. Рискуя прослыть придирчивыми, мы должны сказать, что еще меньше доступной информации имеется о нарушениях слуха при многих муколипидозах и сфинголипидозах. Обычно недостаток этих сведений является следствием того, что указанные заболевания часто сопровождаются глубоким слабоумием и ранней глухотой. Болезнь Тея — Сакса, согласно данным Kelemen, представившего обзор литературы и изучившего лично 2 случая, нередко сопровождается гиперакузией и средним отитом. Это выдержка из работы Boies. У больных GM1-ганглиозидозом, тип I, наблюдается нейросенсорная глухота (R. Desnick, личное сообщение). Goldberg с соавт. обнаружили двустороннюю нейросенсорную глухоту при болезни накопления, характеризующейся задержкой роста, грубыми чертами лица, умственной отсталостью, судорожными припадками, помутнением роговицы, вишнево-красным пятном на глазном дне, множественными дизостозами, дефицитом р-галактозидазы и аутосомно-рецессивным наследованием. Согласно нашим наблюдениям, у больных с муколипидозом-III часто выявляется легкая проводящая глухота.
Молочная ацидемия, миопатия и низкий рост с глухотойHackett, Bray, Ziter, Nyhan и Creer описали 2 сестер с этим, несомненно уникальным, синдромом. У обеих сестер отмечался низкий рост (ниже 3 S.D.). Нервная система. Интеллект был нормальным. У одной из сестер проявилась светостимулирующаяся эпилепсия. Орган слуха. У обеих сестер отмечалась умеренная нейросенсорная глухота, более подробно не охарактеризованная. Лабораторные данные. В моче и в крови у больных было обнаружено высокое содержание аланина. Нагрузочная проба с пищевым введением аланина выявила снижение почечного клиренса аланина. В крови отмечалось заметное повышение уровня пировиноградной и молочной кислот. В моче обнаружено низкое содержание креатина, но уровень креатин-фосфокиназы в сыворотке крови был нормальным. Патология. При люминесцентной микроскопии в скелетных и в сердечной мышцах были обнаружены очаги «гранулярного некроза». Ультраструктурное исследование этих областей выявило многочисленные большие митохондрии с дегенеративно измененными миофибриллами (D’Agostino et al.). Диагноз. Это заболевание имеет некоторое сходство с синдромом лрогрессирующей наружной офтальмоплегии с пигментной дегенерацией сетчатки, дефектом сердечной проводимости и смешанной глухотой и несколькими заболеваниями, обсуждаемыми в диагностическом плане с этим синдромом. Прогноз. Прогноз, несомненно, неблагоприятный. У одной из сестер молочный ацидоз привел к летальному исходу. Выводы. Синдром характеризуется: – Также рекомендуем “Гиперпролинемия и гиперпролинурия (иминоглицинурия) с глухотой” Оглавление темы “Наследственные болезни с глухотой”:
|
Источник
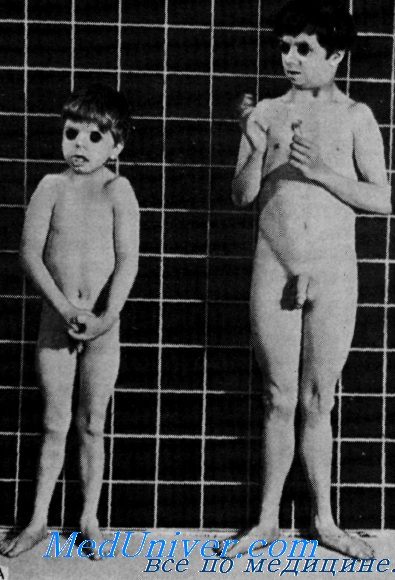
Пренатальное нарушение роста, с повышенным уровнем гормона роста и умственная отсталость с глухотойVan Gemund, Laurent de Angulo и van Gelderen сообщили о 2 мальчиках сибсах с пренаталыюй карликовостью, повышением в сыворотке крови иммунореактивного гормона роста и отсутствием реакции на пего организма, умственной отсталостью и врожденной глухотой. Клинические данные. Данные осмотра. У сибсов отмечалась внутриутробная задержка роста (при рождении в срок вес был менее чем 1900 г), но конечности и туловище были пропорциональными. Голова была несколько малых размеров. Признаки полового развития не выявлялись до 20-летнего возраста, даже под влиянием терапии. Орган слуха. У обоих сибсов была врожденная глухота и плохо развитая речь. Нарушения слуха подробно не описаны. Лабораторные данные. Рентгенологическое исследование показало резкую задержку созревания скелета. В сыворотке крови было обнаружено повышение уровня иммупореактивного гормона роста. Введенный экзогенно человеческий гормон роста вызывал нормальную реакцию в сыворотке крови уровней инсулина, глюкозы и свободных жиров, но недостаточное увеличение ретенции азота и выделения с мочой гидроксипролина.
Наследственность. Родители 2 больных мальчиков были здоровы и состояли в кровно-родственном браке. Заболевание, по-видимому, наследуется по аутосомно-рецессивному типу. Диагноз. Laron описал карликовость с высоким содержанием в сыворотке крови иммупореактивного человеческого гормона роста. На экзогенное введение гормона роста обследованные лица реагировали нормально. Najjar сообщил о сибсах, у которых ни при пероральном, ни при внутривенном введении глюкозы уровень человеческого гормона роста не снижался. Экзогенный гормон роста недостаточно повышал ретенцию азота и экскрецию с мочой гидроксипролина. Ни в одной семье не отмечалось умственной отсталости или глухоты. Лечение. Росту может способствовать терапия анаболическими стероидами. Прогноз. Хотя заболевание не угрожает жизни, прогноз неблагоприятный, так как лечение неэффективно. Выводы. Характеристика этого синдрома включает: – Также рекомендуем “Гипоталамо-гипофизарная карликовость с глухотой. Мукополисахаридозы: синдром Гурлер” Оглавление темы “Наследственные болезни с глухотой”:
|
Источник
Синдромы Хантер, Санфилиппо, Моркио, Марото — Лами. Орган слуха при мукополисахаридозахСиндром Хантер (МПС-II) встречается в виде легкой формы (тип А) и в виде тяжелой формы (тип Б). При типе А больные доживают до взрослого возраста. При типе Б быстро наступают психомоторные расстройства, и больные обычно гибнут до наступления пубертатного возраста. При типе А отмечаются легкие интеллектуальные нарушения. Рост задержан менее резко, чем при МПС-1-Г. В противоположность больным с синдромом Гурлер у детей с синдромом Хаптер обычно не наблюдается выраженного, бросающегося в глаза помутнения роговицы. Синдром Санфилиппо (МПС-III) встречается в виде двух неаллельных форм. Изменения лица у больных выражены значительно менее резко, чем при синдроме МПС-1-Г, роговицы чистые, рост почти нормальный. В дошкольном возрасте дети становятся возбужденными и агрессивными. Смерть наступает обычно во втором десятилетии жизни. Синдром Моркио (МПС – IV) характеризуется развивающимся со второго года жизни выраженным нарушением роста, короткой шеей, «птичьей грудью», прогрессирующей деформацией позвоночника и другими скелетными аномалиями, такими, как вальгусная деформация коленей и плоскостопие. Лицо нормальное, конечности диспропорционально длинные. Почти постоянно наблюдается чрезмерная подвижность суставов. Интеллект почти всегда нормальный. Синдром Марото — Лами (МПС-VI). У больных отмечается резко выраженная гурлсроподобиая внешность, но интеллект нормальный. Существуют две формы — легкая и тяжелая. При легкой форме Характерные изменения впервые выявляются примерно в 6-летнем возрасте, когда замечают маленький рост и изменения со стороны позвоночника. Эти больные доживают до зрелого возраста. Тяжелая форма определяется уже в раннем детстве на основании резко выраженных изменений лица и скелета, выраженного дефекта зрения, потери слуха и порока сердца, ведущего к гибели больных в юности. Орган слуха. Это, вероятно, наиболее трудный для написания раздел во всей книге, так как фактически твердых, достоверных данных о нарушениях слуха при различных типах мукополисахаридозов не имеется. Хотя имеются ранние исследования 75 случаев, представляющие смешанную группу больных мукополисахаридозами. У 25% из них были обнаружены нарушения слуха или глухота. Этот процент ни в коей мерс не отражает частоту дефекта слуха при каждой форме (Ricci, Ancetti, Kittel). Поэтому мы прочитали более 200 сообщений о различных типах нарушений слуха. Такой подход, очевидно, не является удовлетворительным, так как умственная отсталость, нарушение контакта и ранняя глухота имеют точно совпадающие черты. Во многих опубликованных случаях не упоминается о глухоте либо потому, что это не входило в цель исследования, либо потому, что не было замечено, так как дефект слуха был минимальным. Следовательно, за редким исключением, большая часть наших заключений являлась просто впечатлением. Мы надеемся, что это признание в невежестве послужит для благих намерений.
У большинства больных с синдромом Гурлер, вероятно, отмечается легкий прогрессирующий дефект слуха по проводящему типу. Деформация носоглотки и повышенная восприимчивость к заболеваниям верхних дыхательных путей ведут к развитию воспаления в среднем ухе. Несмотря на то что большинство исследователей описывают проводящую потерю слуха, только Kelemen сообщает о патологических изменениях височной кости. У его больного отмечался дефект слуха, равный 60 дБ, главным образом в диапазоне средних частот. Воздушные клетки tegmen и epitympanum были заполнены ретикулярной ме зенхимальной тканью. Слизистая оболочка среднего уха была утолщена и папилломатозна, ниши овального и круглого окон, расположенные в костных разрастаниях, были замурованы. Эти изменения рассматривались как пренатальное проявление заболевания. При синдроме Шейe систематические аудиометрические исследования не проводились. Согласно данным наших таблиц, нарушения слуха были обнаружены, возможно, не более чем у 10—20% больных. Они обычно были выражены не резко и выявлялись в среднем возрасте. Хотя адекватной документации и не имеется, тип нарушений слуха был, по-видимому, смешанным (Koskenoja, Suvanto, Murray, Scheie et al.). Синдром Хантера примерно в половине случаев сопровождается глухотой, хотя дефект слуха обычно не глубокий (Leroy, Crocker). Несмотря на то что ряд исследователей утверждают, что при синдроме Хантер имеются нарушения слуха нейросенсорного типа, на основании наших ограниченных исследований мы предполагаем, что при этом заболевании дефект слуха чаще смешанный. Kittel проиллюстрировал смешанную глухоту у своего больного. Wolff не обнаружила сустава между молоточком и наковальней. Она не описала типичные отосклеротические участки в области круглого окна, но отмстила нерегулярные костные узелки, выпячивающиеся в самую нижнюю часть барабанной лестницы, а также изменения, напоминающие те, о которых сообщил Kelemen при синдроме Гурлср. Отмечались также изменения в кортиевом органе, но они выглядели как посмертные артефакты. Zechner и Altmann нашли в среднем ухе отечную слизистую оболочку, содержащую большие пенящиеся клетки с ПАСК-положительной цитоплазмой. Молоточек и наковальня были нормальной формы, но содержали большие костномозговые полости, наполненные кровью. Стремя и система воздушных клеток выглядели нормальными. ПАСК-положительные клетки были обнаружены внутри воздушных клеток сосцевидного отростка, и толстый ПАСК-положительпый плащ окружал мелкие кровеносные сосуды. В области полукружных каналов были видны многочисленные голубые плащи. В области овального и круглого окон обнаружены отосклеротические фокусы. Кортиев орган был нормальным. В нижней части сосудистой полости отмечалась широкая ПАСК-положитсльная зона. Цитоплазма спирального и вестибулярного узлов была заполнена пенящимся ПАСК-положительным веществом. При синдроме Санфилиппо очень редко сообщают о нарушениях слуха. Дефект слуха был обнаружен у 1 из 10 больных, описанных Spranger с сотр., и у 2 или, возможно, 3 больных из 8, изученных Rampini. Однако агрессивное поведение и нарушение контакта, наблюдающиеся у этих больных, создавали трудности вплоть до полной невозможности проведения аудиометрических исследований. Единичные данные позволяют предположить, что, когда при этом заболевании имеется дефект слуха, он выявляется в 6—7-летнем возрасте и в дальнейшем прогрессирует. У большей части больных с синдромом Моркио наблюдается смешанная глухота. Выявляется она обычно в течение второго десятилетия жизни и в большинстве случаев не достигает резко выраженной степени (Robins et al, Von Noorden et al.). Нарушения слуха, по-видимому, проводящего типа отмечаются почти у 25% больных с синдромом Марото — Лами. Выявляются они в 6—8 лет и развиваются в результате часто повторяющихся средних отитов (Liebenam, Stocckel, Maroteaux et al., Sarrouy et al., 1965; Fallis et al., Spranger et al.). Вестибулярная система. Единственное исследование, имеющее отношение к больному с синдромом Хантера, в котором описана вестибулярная функция, указывает на двустороннее угнетение реакций. – Также рекомендуем “Диагностика, патология, наследственность и течение мукополисахаридозов” Оглавление темы “Наследственные болезни с глухотой”:
|
Источник