
Синдром эдвардса по мкб 10

Содержание
- Описание
- Симптомы
- Диагностика
- Лечение
Названия
Синдром Эдвардса.
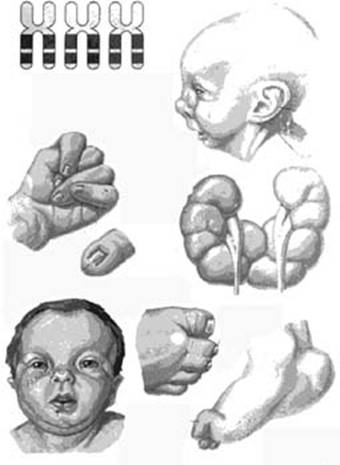
Симптомы при синдроме Эдвардса (трисомия 18)
Описание
Впервые трисомию по группе Е описали J. Edwads и соавт. (1960г. ). Частота синдрома Эдвардса среди новорожденных в среднем составляет 1:7000, замечено, что девочки поражаются в 3 раза чаще, чем мальчики. Ученым высказано предположение о стабилизирующем действии Х-хромосомы при абберациях пары 18, тогда как зиготы с трисомией 18, имеющие мужской генотип, элиминируются. Средний возраст матерей 32,5 года, отцов – 35 лет.
Симптомы
Длительность беременности превышает нормальную, и составляет в среднем 42 недели. Во время беременности диагностируют слабую активность плода и многоводие. Отмечается,что плацента имеет меньшие, чем обычно, размеры, часто обнаруживается только одна пупочная артерия; многие дети рождаются в асфиксии, с очень низкой массой тела и выраженной гипотрофией.
Фенотип больных с синдромом Эдвардса довольно характерен. Череп долихоцефалический, с низким лбом, затылок более широкий и выступающий. Часто встречается микроцефалия или гидроцефалия. Надглазничные валики сглажены, глазные щели узкие, наблюдается эпикант, птоз, часто встречается глазная патология, микрофтальмия, колобома, катаракта. Переносье вдавлено, но спинка носа тонкая, выступающая, ушные раковины расположены очень низко, диагностируется отсутствие мочки и козелка, недоразвиты завиток и противозавиток. Характерна микроретрогнатия. Рот мешьше обычных размеров, имеет треугольную форму, верхняя губа короче обычного. Небо высокое, иногда с расщелиной, шея короткая, нередко с крыловидной складкой.
Аномалии опорно-двигательного аппарата отличаются разнообразием: расширение шрудной клетки, укорочение грудины, таз узкий, деформации конечностей, ограничена подвижность в тазобедренных суставах, описаны вывихи бедра. Кисти и пальцы короткие, клинодактилия 5 пальцев кисти, дистально расположенный, гипоплазированный 1 палец кисти, сглажен тенар. Пальцы сжаты в кулак по типу «флексорной аномалии»: 2 и 5 пальцы расположены сверху и прикрывают прижатые к ладони 3 и 4 пальцы; 1 палец стопы широкий и короткий, синдактилия 2 и 3 пальцев. Типична для трисомии 18 форма стопы в виде своеобразной «качалки». Характерна общая мышечная гипотония. У мальчиков нередко встречается крипторхизм, гипоспадия; гипертрофия клитора у девочек.
Интеллектуальный дефект соответствует олигофрении в степени идиотии ил глубокой имбецильности и только в единичных описаниях мозаичного варианта трисомии 18 умственное недоразвитие менее грубое. Нередко у больного развиваются судороги.
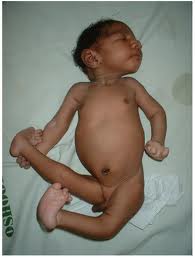
Внешний вид больного с синдромом Эдвардса (трисомия 18)
Диагностика
Дерматографическая картина при синдроме Эдвардса отличается своеобразными признаками: отмечается большая частота дуг на подушечках пальцев рук (примерно в 10 раз выше, чем в общей популяции), нередко дистальная сгибательная складка на пальцах не развита, у трети больных обнаруживается поперечная ладонная борозда, гребневый счет увеличен, осевой трирадиус обычно расположен дистально.
При аутопсии при синдроме Эдвардса находят разнообразные пороки развития практически всех органов и систем. С различной частотой встречаются аномалии ЦНС: аплазия или гипоплазия мозолистого тела, гипоплазия мозжечка, атрофия мозговых извилин. У 95% пациентов с трисомией 18 диагностируются пороки сердца и крупных сосудов, наиболее часто встречаются дефект межжелудочковой перегородки и незаращение артериального протока. Около половины всех случаев синдрома Эдвардса сопровождается аномалиями органов пищеварения: нарушение расположения кишечника, меккелев дивертикул, атрезия пищевода, атрезия заднего прохода. В 50% случаев отмечаются пороки развития мочеполовой системы – дольчатая или подковообразная почка, дупликация мочеточников, гипоплазия яичников.
При цитогенетическом обследовании в 80% случаев находят трисомию 18, у 10% больных – мозайцизм. Описаны случаи транслокационного варианта, двойной анеуплодии типа 48, ХХУ +18 с участием трисомного по хромосоме 18 клона.
Лечение
Нет патогенетического лечения синдрома Эдвардса на сегодняшний день, возможна лишь симптоматическая коррекция патологий. Прогноз для жизни неблагоприятный, средняя продолжительность жизни мальчиков 2-3 месяца, девочек – 10 месяцев. 30% больных умирают в течение 1-го года жизни, до года доживает лишь 10% больных. При мозаичных вариантах прогноз для жизни несколько лучше.
Источник
Рубрика МКБ-10: Q91.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q90-Q99 Хромосомные аномалии, не классифицированные в других рубриках / Q91 Синдром Эдвардса и синдром Патау
Определение и общие сведения[править]
Cиндром Эдвардса
Синонимы: трисомия 18
Трисомия по 18-й хромосоме встречается у новорожденных с частотой от 1:3300 до 1:10 000; у девочек бывает в 3 раза чаще, чем у мальчиков. Больные дети часто рождаются недоношенными или переношенными. Нарушения при трисомии по 18-й хромосоме гораздо тяжелее, чем при синдроме Дауна; лишь 50% пробандов доживают до 2-месячного возраста; 10% живут 1 год. Средняя продолжительность жизни мальчиков — 60, девочек — 280 дней.
Этиология и патогенез[править]
Большинство случаев связаны со свободной трисомией 18. Мозаичная трисомия 18 была обнаружена у нескольких пациентов с клинической картиной, которая варьируется от классической трисомии 18 до нормального фенотипа в зависимости от количества трисомных клеток, присутствующих в тканях. Фенотип трисомии 18, по-видимому, связан с наличием трех копий интервала 18q11-q12.
Клинические проявления[править]
Клиническая картина: череп необычной формы (узкий лоб и широкий выступающий затылок), низкое расположение ушей, микрогнатия, сгибательная контрактура кистей и стоп, дисплазия стоп, пороки сердца, сильная задержка психического развития. Главные нарушения обмена веществ и эндокринные расстройства: гипоплазия подкожной клетчатки, сильная задержка роста. Дисгенезия щитовидной железы или надпочечников встречается менее чем у 10% больных.
Синдром Эдвардса неуточненный: Диагностика[править]
Трисомию 18 можно заподозрить во время беременности по результатам ультразвукового исследования плода (задержка роста, пороки развития, множественные кисты сосудистого сплетения) и подтверждить кариотипическим анализом плода. Маркеры сыворотки (используемые для диагностики трисомии 21) также могут быть аномальными.
Дифференциальный диагноз[править]
Синдром Эдвардса неуточненный: Лечение[править]
Хирургическое лечение пороков развития мало способствует улучшению неблагоприятного прогноза, связанного с этим синдромом: 90% детей умирают в течение первого года жизни от сердечных, почечных или неврологических осложнений или от повторных инфекций.
Сообщалось о длительном выживании (в некоторых случаях до взрослого возраста), главным образом в случаях мозаичной или частичной трисомии (в результате транслокации). Большинство немозаичных пациентов имеют лишь ограниченную автономию (отсутствие речи и ходьбы).
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Дополнительная литература (рекомендуемая)[править]
1. Buyse ML. Birth Defects Encyclopedia. Cambridge: Blackwell, 1990.
2. Emery A, Rimoin D. Principles and Practice of Medical Genetics (2nd ed). New York: Churchill Livingstone, 1990.
3. Gorlin RJ, et al. Syndromes of the Head and Neck (3rd ed). New York: Oxford University Press, 1990.
4. Hall JG, et al. Handbook of Normal Physical Measurements. New York: Oxford University Press, 1989.
5. Jones KL. Smith’s Recognizable Patterns of Human Malformation (4th ed). In M Markowitz (ed.), Major Problems in Clinical Pediatrics (vol VII). Philadelphia: Saunders, 1988.
6. McKusick V. Mendelian Inheritance in Man (10th ed.). Baltimore: Johns Hopkins University Press, 1992.
7. Rimoin DL. Disorders of the Endocrine Glands. In AA Dietz (ed.), Genetic Disease: Diagnosis and Treatment. Proceedings of the Fifth Arnold O. Beckman Conference in Clinical Chemistry. Monterey, CA: The Association for Clinical Chemistry, 1983.
8. Taybi H, Lachman RS. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasias (3rd ed). Chicago: Yearbook, 1990.
9. Vogel F, Motulsky AG. Human Genetics: Problems and Approaches (2nd ed). New York: Springer, 1986.
10. Wiedmann H-R, et al. Atlas of Clinical Syndromes—A Visual Aid to Diagnosis (2nd ed). St. Louis: Mosby, 1989.
11. Wynne-Davis R, Hall CM, Apley AG. Atlas of Skeletal Dysplasias. Edinburgh: Churchill Livingstone, 1985.
Действующие вещества[править]
Источник

Синдром Эдвардса – второе по распространенности генетическое заболевание после синдрома Дауна, связанное с хромосомными аберрациями. При синдроме Эдвардса наблюдается полная или частичная трисомия 18-й хромосомы, в результате которой образуется ее лишняя копия. Это провоцирует ряд необратимых нарушений организма, которые в большинстве случаев несовместимы с жизнью. Частота встречаемости данной патологии составляет один случай на 5-7 тысяч детей, при этом большинство новорожденных с симптомом Эдвардса – девочки. Исследователи предполагает, что дети мужского пола погибают еще в перинатальный период либо в процессе родов.
Впервые описал заболевание в 1960 году генетик Эдвардс, который выделил более 130 симптомов, характеризующих данную патологию. Синдром Эдвардса не наследуется, а является результатом мутации, вероятность возникновения которой составляет 1%. Факторы, провоцирующие патологию – радиационное воздействие, кровное родство между отцом и матерью, хроническое воздействие никотина и алкоголя в период зачатия и беременности, контакт с химически агрессивными веществами.
Синдром Эдвардса – генетическое заболевания, связанное с неправильным делением хромосом, из-за которого образуется лишняя копия 18-й хромосомы. Это приводит к ряду генетических нарушений, что проявляется серьезными патологиями организма как умственная отсталость, врожденные пороки сердца, печени, ЦНС, опорно-двигательного аппарата.
Встречаемость у заболевания достаточно редкая – 1:7000 случаев, при этом большинство новорожденных с синдромом Эдвардса не доживают до первого года жизни. Среди взрослых пациентов большинство (75%) составляют женщины, так как зародыши мужского пола, обладающие данной патологией, погибают еще в период внутриутробного развития, из-за чего беременность заканчивается выкидышем.
Главным фактором риска для развития синдрома Эдвардса является возраст матери, так как нерасхождение хромосом, являющееся причиной патологии плода, в большинстве случаев (90%) происходит у материнской половой клетки. Остальные 10% случаев синдрома Эдвардса связаны с транслокациями и нерасхождением хромосом зиготы во время дробления.
Синдром Эдвардса, как и синдром Дауна чаще встречается у детей, матери которых забеременели в возрасте старше сорока. (читайте также: Причины и симптомы синдрома дауна)
Для оказания своевременной медицинской помощи детям с врожденными пороками развития, которые спровоцированы хромосомными аномалиями, новорожденные должны проходить обследование у кардиолога, невролога, детского уролога и ортопеда. Сразу же после рождения младенцу необходимо диагностическое обследование, которое включает УЗИ малого таза и брюшной полости, а также эхокардиография для выявления нарушений сердечной деятельности.
Содержание:
- Симптомы синдрома Эдвардса
- Причины синдрома Эдвардса
- Диагностика синдрома Эдвардса
- Лечение синдрома Эдвардса
Симптомы синдрома Эдвардса
Патологическое течение беременности является одним из главных признаков наличия синдрома Эдвардса. Плод малоактивен, недостаточный размер плаценты, многоводие, только одна пупочная артерия. При рождении младенцы с синдромом Эдвардса отличаются низкой массой тела, даже если беременность была переношенной, асфиксия сразу после рождения.
Ряд врожденных патологий младенцев с синдромом Эдвардса приводит к тому, что большинство из них погибают в первые недели жизни из-за проблем с сердцем, невозможности нормального дыхания и пищеварения. Сразу после рождения их питание осуществляется через зонд, так как они не могут сосать и глотать, возникает необходимость искусственной вентиляции легких.
Большинство симптомов заметны невооруженным взглядом, поэтому заболевание диагностируют практически сразу. К внешним проявлениям синдрома Эдвардса относятся: укороченная грудина, косолапость, вывих бедра и аномальное строение ребер, скрещенные пальцы кистей рук, кожа покрыта папилломами или гемангиомами. Кроме того, у новорожденных с данной патологией специфическое строение лица – низкий лоб, укороченная шея с излишней кожной складкой, маленький рот, заячья губа, выпуклый затылок и микрофтальмия; уши расположены низко, слуховые проходы слишком узкие, ушные раковины деформированы.
У детей с синдромом Эдвардса наблюдаются серьезные нарушения ЦНС – микроцефалия, гипоплазия мозжечка, гидроцефалия, менингомиелоцеле и другие. Все эти пороки развития приводят к нарушению интеллекта, олигофрении, глубокой идиотии.
Симптомы синдрома Эдвардса разнообразны, заболевание имеет проявления со стороны практически всех систем и органов – поражения аорты, перегородок сердца и клапанов, кишечная непроходимость, пищеводные свищи, пупочные и паховые грыжи. Со стороны мочеполовой системы у младенцев мужского пола часто встречается неопущение яичек, у девочек – гипертрофия клитора и двурогая матка, а также общие патологии – гидронефроз, почечная недостаточность, дивертикулы мочевого пузыря.
Причины синдрома Эдвардса
Хромосомные нарушения, которые приводят к возникновению синдрома Эдвардса, происходят еще на стадии формирования половых клеток – овогенеза и сперматогенеза, либо появляются при неправильном дроблении зиготы, образованной двумя половыми клетками.
Риски возникновения синдрома Эдварда те же, что и для других хромосомных аномалий, во многом совпадают с таковыми для синдрома Дауна.
Вероятность возникновения патологии возрастает под воздействием некоторых факторов, в числе которых одним из главных считается возраст матери. Частота встречаемости синдрома Эдвардса выше у женщин, которые рожают в возрасте старше 45. К хромосомным аномалиям приводит воздействие радиационного излучения, также этому способствует хроническое употребление алкоголя, наркотических веществ, сильнодействующих лекарственных препаратов, курение. Воздерживаться от вредных привычек и избегать влияния химически агрессивных веществ на рабочем месте или регионе проживания рекомендуется не только в период беременности, но и несколько месяцев до зачатия.
Диагностика синдрома Эдвардса
Своевременная диагностика позволяет выявить хромосомное нарушение на ранних этапах беременности и принять решение о целесообразности ее сохранения, с учетом всех возможных осложнений и врожденных пороков плода. Ультразвуковое исследование у беременных не дает достаточно данных для диагностирования синдрома Эдвардса и других генетических заболеваний, но может дать информацию о ходе беременности. Отклонения от нормы, такие как многоводие или малый размер плода, дают основания для проведения дополнительных исследований, включения женщины в группу риска и усиленного контроля над ходом беременности в дальнейшем.
Пренатальный скрининг – эффективная диагностическая процедура, позволяющая обнаружить пороки развития на ранней стадии. Скрининг проходит в два этапа, первый из которых проводится на 11-й неделе беременности и заключается в исследовании биохимических показателей крови. Данные об угрозе синдрома Эдвардса на первом триместре беременности не являются окончательными, чтобы подтвердить их достоверность необходимо пройти второй этап скрининга
Женщинам, которые попали в группу риска синдрома Эдвардса, рекомендуют пройти инвазивное обследование для подтверждения диагноза, что помогает разработать дальнейшую стратегию поведения.
Другие признаки, указывающие на развитие синдрома Эдвардса – аномалии развития плода, обнаруженные на УЗИ, обилие околоплодных вод при малом размере плаценты, агенезия пупочной артерии. Помочь в диагностике синдрома Эдвардса могут данные допплерографии маточно-плацентарного кровообращения, УЗИ и стандартного скрининга.
Помимо показателей состояния плода и патологического хода беременностями, основаниями для зачисления будущей матери в группу повышенного риска является возраст старше 40-45 и избыточный вес.
Для определения состояния плода и особенностей протекания беременности на первом этапе скрининга необходимо получить данные о концентрации белка PAPP-A и бета-субъединиц хорионического гонадотропина (ХГЧ). ХГЧ вырабатывается самим эмбрионом, а по мере его развития – плацентой, окружающей плод.
Второй этап проводится начиная с 20-й недели беременности, включает отбор образцов тканей для гистологических исследований. Для этих целей лучше всего подходят пуповинная кровь и околоплодная жидкость. На данной стадии перинатального скрининга можно с достаточной точностью сделать выводы о кариотипе ребенка. Если результат исследования отрицательный, то хромосомных аномалий нет, в противном случае существуют основания для постановки диагноза синдрома Эдвардса.
Лечение синдрома Эдвардса

Как и при других генетических заболеваниях, вызванных хромосомными аномалиями, прогноз для детей с синдромом Эдвардса неутешительный. Многие из них погибают сразу при рождении или в течение нескольких дней, несмотря на оказанную медицинскую помощь. Девочки могут прожить до десяти месяцев, мальчики погибают в течение первых двух-трех. Только 1% новорожденных доживает до десятилетнего возраста, при этом о самостоятельности и социальной адаптации не может быть и речи из-за серьезных нарушений интеллекта.
Больше шансов выжить в первые месяцы у пациентов с мозаичной формой синдрома, так как повреждения касаются не всех клеток организма. Мозаичная форма встречается в случае, если хромосомные аномалии произошли на этапе деления зиготы, уже после слияния мужской и женской половых клеток. Тогда та клетка, в которой было нерасхождение хромосом, из-за чего образовалась трисомия, при делении дает начало аномальным клеткам, что и провоцирует все патологические явления. Если же трисомия произошла на этапе гаметогенеза с одной из половых клеток, то аномальными будут все клетки плода.
Лекарственного средства, которое могло бы повысить шансы на выздоровление, не существует, так как пока не представляется возможным вмешательство на хромосомном уровне во всех клетках организма. Единственное, что может предложить современная медицина – симптоматическое лечение и поддержание жизнеспособности ребенка. Коррекция патологических явлений, связанных с синдромом Эдвардса, может улучшить качество жизни пациента, продлить ее срок. Хирургическое вмешательство при врожденных пороках развития нецелесообразно, так как влечет большие риски для жизни пациента и имеет множество осложнений.
Больные с синдромом Эдвардса с первых дней жизни должны наблюдаться у педиатра, так как они очень уязвимы для возбудителей инфекции. Среди новорожденных с данной патологией часто встречаются конъюнктивит, инфекционные заболевания мочеполовой системы, средний отит, синусит, пневмония.
Родителей ребенка с синдромом Эдвардса часто беспокоит вопрос – можно ли рожать еще раз, какова вероятность того, что следующая беременность также будет патологической. Исследования подтверждают, что риск повторного явления синдрома Эдвардса у одной и той же супружеской пары очень низок, даже в сравнении со средней вероятностью в 1% случаев. Вероятность рождение следующего ребенка с той же патологией составляет примерно 0,01%.
В целях своевременной диагностики синдрома Эдвардса будущим матерям рекомендуется проводить антенатальный скрининг в период беременности. При обнаружении патологий на ранних стадиях беременности можно будет сделать аборт по медицинским показаниям.
Автор статьи: Мочалов Павел Александрович | д. м. н. терапевт
Образование:
Московский медицинский институт им. И. М. Сеченова, специальность – “Лечебное дело” в 1991 году, в 1993 году “Профессиональные болезни”, в 1996 году “Терапия”.
Наши авторы
Источник