
Синдром элерса данло частота встречаемости

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 26 июля 2015; проверки требуют 20 правок.
Синдром Э́лерса — Данло́са (синдром Э́лерса — Данло́[2], син. Э-Д; англ. Ehlers-Danlos Syndrome, «гиперэластичность кожи» («Cutis hyperelastica»[3]), несовершенный десмогенез, несовершенный десмогенез Русакова, синдром Черногубова — Элерса — Данлоса) — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.
Синдром назван в честь двух дерматологов, идентифицировавших его в начале XX века: Эдварда Элерса[en] (1863—1937) из Дании и Анри-Александра Данлоса[fr] (1844—1912) из Франции[4].
Симптоматика[править | править код]
Человек с син. Э-Д демонстрирует гиперподвижность суставов. |
Симптомы сильно варьируются в зависимости от типа болезни. Болезнь, как правило, поражает суставы, кожу и кровеносные сосуды, с симптомами, такими как свободные (плохо прикреплённые), сильно гнущиеся суставы; гладкая или эластичная, легко повреждающаяся кожа; неправильное заживление ран и формирование шрамов; маленькие и хрупкие кровеносные сосуды. Все формы затрагивают суставы, вызывая гиперподвижность, они выходят за пределы нормального диапазона движений.[5] В результате люди с «син. Э-Д» более подвержены различным травмам таким как: вывихи, подвывихи, растяжения связок, деформация и иногда разрыв мягких тканей. Так как синдром часто не диагностируется, некоторые случаи принимаются за жестокое обращение с детьми.[6]
Классификация[править | править код]
В прошлом было более десяти общепризнанных типов «син. Э-Д». В 1997 исследователи предложили более простую классификацию, которая сократила число главных типов до шести и дала им описательные имена.[7] Другие типы условно могут существовать, но о них сообщается только в отдельных семьях или они плохо характеризованы. За исключением типа гиперподвижности, были идентифицированы и отождествлены отдельные мутации — они могут быть точно определены генетическим анализом. Это особенно важно из-за значительной вариативности признаков в индивидуальном рассмотрении, из-за чего может быть перепутана классификация только на симптоматической основе.
По распространенности среди населения:
| Имя | Номер | Описание | OMIM | Ген(ы) |
| Гиперподвижность (Hypermobility) | тип 3 | Поражает 1 человека на 10 000 – 15 000, вызван аутосомным доминирующим механизмом. Возникает в результате мутации любого из двух генов, которые вызывают Сосудистый тип и син. Э-Д с дефицитом тенасцина-X, соответственно. Проявляется в гиперподвижности суставов; поражение кожи выражено меньше. Характерны хронические мышечно-скелетные боли. | 130020 | COL3A1, TNXB |
| Классический (Classical) | тип 1 и 2 | Поражает приблизительно от 2 до 5 человек на 100 000. Затрагивает коллаген типа V, также коллаген типа I. | 130000, 130010 | COL5A1, COL5A2, COL1A1 |
| Сосудистый (Vascular) | тип 4 | Поражает приблизительно 1 человека на 100 000. Вызван аутосомно-доминантным дефектом в синтезе коллагена типа III | 130050 | COL3A1 |
| Кифосколиоз (Kyphoscoliosis) | тип 6 | Аутосомно-доминантный дефект, вызывающий недостаток фермента, называемого Лизин Гидролаза[1]. Очень редок; описано немногим более 60 случаев. | 225400, 229200 | PLOD1 |
| Артрохалазия (Arthrochalasis) | типы 7A и B | Поражает коллаген типа I. Крайне редок, описано всего около 30 случаев. | 130060 | COL1A1, COL1A2 |
| Дерматоспараксис (Dermatosparaxis) | тип 7C | Также крайне редок, описано около 10 случаев. | 225410 | ADAMTS2 |
Тип гиперподвижность[править | править код]
Прежний тип 3, встречается у 1 человека на 10 000 — 15 000, что делает его самым распространённым вариантом болезни. Признаки и симптомы могут быть не диагностированы (не признаны) врачами или, как правило, ошибочно диагностированы как фибромиалгия и обычно больным не ставят диагноз, пока не проявятся серьёзные осложнения. Диагностика осуществляется, в основном, на клинических наблюдениях.
Основные признаки и симптомы включают в себя:
- Свободные, нестабильные суставы, подверженные: растяжениям, вывихам, подвывихам (частичный вывих), переразгибанию суставов
- Плоскостопие
- Высокое и узкое нёбо
- Лёгкие кровоподтёки
- Легко повреждающаяся бархатно-гладкая кожа
- Раннее начало остеопороза (обычно проявляется в середине 30 лет)
- Поражение сердца: в некоторой степени en:Dysautonomia или приобретённый порок сердца (такой как пролапс митрального клапана, что создаёт повышенный риск инфекции (эндокардит) во время операций (также возможно развитие до крайне опасной для жизни степени при пролапсе митрального клапана).[8]
Другие симптомы и осложнения могут включать в себя:
- Низкая плотность костей (остеопения) — предшественник остеопороза
- Мышечная слабость, часто усугубляется холодной погодой
- Деформации позвоночника, такие как: сколиоз (искривление позвоночника), кифоз (горб в грудном отделе), en:Tethered spinal cord syndrome, базилярная инвагинация (cranial settling), а также Мальформация Арнольда — Киари (поражение продолговатого мозга, мозжечка выраженные затылочными болями, нарушениями глотания, атаксией и другими симптомами)[9]
- Функциональные расстройства кишечника (функциональный гастрит, синдром раздражённого кишечника)
- Сдавление нервов (синдром запястного канала, парестезия, невралгия тройничного нерва)
- Болезнь Рейно
- Миалгия (боль в мышцах) и артралгия
- Чрезмерная усталость
- Преждевременный разрыв амниона (выкидыш) во время беременности en:Premature rupture of membranes
- Младенцы с гипервподвижностью суставов имеют слабый мышечный тонус (мышечная гипотония), который может задержать развитие таких моторных навыков как самостоятельное присаживание, вставание и хождение.
Боль, сопутствующая этому состоянию, является серьёзным осложнением.
Классический тип[править | править код]
При старой системе классификации он был разделён на два типа: тип I (тяжёлый) и тип II (умеренный). Поражает приблизительно от 2 до 5 человек на 100 000, и является вторым по распространённости. Поражает коллаген V и I типа. Важные симптомы затрагивают кожу и суставы. Больные как правило проявляют:
- гладкая, сильно эластичная, легко ранимая кожа
- уродливые или необычайно обширные шрамы, особенно на лбу, коленях, локтях и подбородке
- гиперподвижные суставы имеют тенденцию к вывихам, растяжению связок и подвывихам (обычно в коленной чашечке, в плече, в пястно-фаланговом суставе en:Metacarpophalangeal joint, и в височно-челюстном суставе
- Из-за сниженного мышечного тонуса, у младенцев может быть нарушено развитие моторных навыков
- Дети могут иметь склонность к развитию грыжи или смешению любого внутреннего органа.
В настоящее время не существует определённого теста для диагностики этого типа синдрома. И ДНК анализ и биохимические исследования используются для выявления пораженных болезнью. В некоторых случаях биопсия кожи была признана полезной при постановке диагноза. К сожалению эти тесты не достаточно надёжны, чтобы выявить всех больных. Если в семье есть несколько больных членов, то можно провести внутриутробную ДНК диагностику.
Сосудистый тип[править | править код]
Поражает приблизительно 1 человека на 100 000, вызван аутосомным доминантным дефектом в синтезе коллагена типа III. В старой системе классификации имел номер IV, этот тип является самой опасной разновидностью синдрома. Проведенные исследования определяют ожидаемую продолжительность жизни примерно в 48 лет. Тем не менее, эта цифра вероятно искажена и основана на факте, что этот тип (как все остальные типы синдрома) значительно не выявлен, и высокая пропорция смертельных случаев вызвана посмертным диагностированием. Повышение осведомлённости среди врачей и населения может помочь сделать эту цифру более точной, и сократить число преждевременных смертей.
Признаки и симптомы:
- Гиперподвижность, наиболее очевидна на пальцах рук и ног
- Хрупкие стенки сосудов оболочек органов и нежной кожи, имеют склонность к разрыву или образованию аневризмы
- Больные как правило имеют тонкую, бледную и прозрачную кожу (можно видеть вены на груди)
- Артериальная/кишечная/маточная хрупкость или трещины, разрывы
- обширные кровоподтёки
- Некоторые пациенты выражают характерные черты лица (большие глаза, маленький подбородок, тонкий нос и губы, мягкие уши) и имеют маленький рост
В результате возможности маточного разрыва, беременность может оказаться опасной для жизни. Доступно лабораторное тестирование. Кожная биопсия может служить доказательством аномальной структуры коллагена. Этот биомеханический анализ выявляет более 95 % случаев. Лабораторное тестирование рекомендуется лицам имеющим два или более значительных симптома. ДНК анализ может выявить изменения в пределах гена COL3A1. Эта информация может помочь при предродовой генетической консультации, когда один из родителей был диагностирован и известна его генетическая мутация или был продемонстрирован биомеханический дефект.
Тип кифосколиоз[править | править код]
В прежней классификации тип VI. Он очень редок, обнаружено немногим более 60 случаев, передаётся аутосомно-рецессивным механизмом. Основным симптом является общая нестабильность (непрочность) суставов. У младенцев наблюдается слабый мышечный тонус, задержка в развитии моторных навыков, прогрессирующее в течение жизни ненормальное искривление позвоночника сколиоз, при котором обычно больные не могут ходить к 20 годам. Легко ранимые глаза и кожа, также вероятна уязвимость кровеносных сосудов. Наблюдаются: спонтанная отслойка сетчатки, кровоизлияния в стекловидное тело, разрывы глазного яблока и роговицы, склер. Также у костей может быть снижена плотность.
Существует четыре основных медицинских критерия при диагностике этого типа. Это гиперподвижные суставы, слабый мышечный тонус у новорождённых, прогрессирующий с рождения сколиоз и хрупкие (уязвимые) глаза, что может придать голубоватый оттенок склерам или вызвать разрыв глаза.
Вызван изменениями в хромосоме 1 гена PLOD, который кодирует фермент лизил-гидролазу. Возможен лабораторный тест, в нём измеряется содержание в моче hydroxylysyl pryridinoline’а. Он крайне чувствителен и специфичен для данного типа синдрома, рекомендуется младенцам с тремя и более основными симптомами. Внутриутробный анализ применяется, если известно о существующем риске: диагностированы больные члены семьи, имеющие положительные результаты тестирования. Вышеупомянутый амниоцентез может быть выполнен, если зародышевые клетки берутся из амниотической жидкости и измерена активность фермента.
Артрохалазия[править | править код]
Характеризуется дефектом коллагена I типа за счет генов COL1.
При этом варианте ребенок может рождаться с врожденным вывихом бедра. Наследуется аутосомно – доминантно. Описано только около 30 случаев.[10]
Симптомы:
- Тяжелая генерализованная гипермобильность суставов с повторными вывихами (подвывихами)
- Гиперэластичная кожа;
- Атрофические шрамы;
- Мышечная гипотония;
- Кифосколиоз;
- Остеопения.[10]
Дерматоспараксис[править | править код]
Имеется дефектный ген ADAMTS2, о котором было сообщено в 10 случаях по всему миру.
Наследуется аутосомно – рецессивно.
Симптомы:
- Чрезвычайно хрупкая кожа, склонная к появлению синяков
- Раны и ссадины тяжело и долго заживают, образуя впоследствии атрофические шрамы
- Кожа на ощупь мягкая и податливая, ее слишком много и она образует складки
- Преждевременный разрыв плодных оболочек, грыжи (пупочные, паховые)[10]
Диагностика[править | править код]
Основана на данных:
- Анамнеза заболевания
- Обследования
- Биопсии кожи
- Молекулярно-генетических исследований
Дополнительно проводят:
- УЗИ внутренних органов
- Офтальмологические обследования
- Эхокардиограмму[10]
Лечение[править | править код]
Симптоматическое:
Для стабилизации деятельности сердечно-сосудистой и нервной системы, нормализации процессов в опорно-двигательном аппарате и коже используются:
- гемостатическая терапия – аскорбиновая кислота, этамзилат, антифибринолигики;
- витаминные препараты (витамины А, Е, В, С);
- препараты, стимулирующие обмен веществ и регенерацию;
- метаболические средства (карнитин-хлорид);
- минеральные комплексы;
- препараты, стимулирующие рост (инъекции соматотропного гормона);
- нейрометаболические стимуляторы, стимулирующие умственную деятельность.[10]
Профилактика[править | править код]
Основными направлениями профилактики неблагоприятных проявлений СЭД являются:
- правильно подобранная физическая нагрузка;
- предотвращение дислокаций;
- профилактические курсы лечения у офтальмолога, стоматолога;
- удаление псевдоопухолей;
- лечение патологии сердца, глаз.
См. также[править | править код]
Дисплазия соединительной ткани
Ссылки[править | править код]
- ↑ база данных Disease ontology (англ.) — 2016.
- ↑ В русскоязычной литературе распространены оба названия, «Синдром Элерса — Данло́са» и «Синдром Элерса — Данло́». Большая медицинская энциклопедия (том 29, 1998), Геномная энциклопедия человека (1991), Малая медицинская энциклопедия (Т. 1, 1991) дают написание «Элерса-Данлоса».
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews’ Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. Page 512. ISBN 0-7216-2921-0.
- ↑ «Uncovered: How U.S. Health System Can Fail Even the Insured — A Woman Endures 16-Month Odyssey To Get a Diagnosis», John Carreyrou, Wall Street Journal, November 16, 2007
- ↑ Lawrence E.J. The clinical presentation of Ehlers-Danlos syndrome (англ.) // Advances in Neonatal Care (англ.)русск. : journal. — 2005. — Vol. 5, no. 6. — P. 301—314. — doi:10.1016/j.adnc.2005.09.006. — PMID 16338669.
- ↑ The Press Enterprise, Redlands mother stung by untrue suspicions presses for accountability in child abuse inquiries Архивировано 28 февраля 2009 года., 2008-04-03
- ↑ Beighton P., De Paepe A., Steinmann B., Tsipouras P., Wenstrup R.J. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) (англ.) // American Journal of Medical Genetics (англ.)русск. : journal. — 1998. — Vol. 77, no. 1. — P. 31—7. — doi:10.1002/(SICI)1096-8628(19980428)77:1<31::AID-AJMG8>3.0.CO;2-O. — PMID 9557891.
- ↑ Ehlers-Danlos Syndrome, Hypermobility Type — GeneReviews — NCBI Bookshelf
- ↑ Sorry, but we cannot find this page. Дата обращения 10 апреля 2013.
- ↑ 1 2 3 4 5 Румянцев А.Г. Федеральные клинические рекомендации по диагностике и лечению синдрома Элерса-Данло у детей (2015).
Источник
- Авторы
- Резюме
- Файлы
- Ключевые слова
- Литература
Макаов А.Х.
1
Ельчинова Г.И.
2
Галкина В.А.
2
Куцев С.И.
2, 3
Зинченко Р.А.
2, 3
1 Муниципальное бюджетное лечебно-профилактическое учреждение «Хабезская центральная районная больница»
2 Федеральное государственное бюджетное научное учреждение «Медико-генетический научный центр»
3 ГБОУ ВПО Российский национальный исследовательский медицинский университет имени Н.И.Пирогова»
Целью данного исследования явился анализ распространенности синдрома Элерса – Данло (СЭД ) на основании популяционного медико-генетического обследования населения ряда популяций России. Проведен анализ распространенности синдрома СЭД в ряде популяций европейской части России: Кировской, Костромской, Тверской, Ростовской, Архангельской, Брянской областей и Краснодарского края, Республик Марий Эл, Удмуртия, Чувашия, Башкортостан, Татария, Адыгея и Карачаево-Черкессия. В большинстве семей клинические проявления различных типов СЭД пересекались, однако можно констатировать, что для всех семей можно было предположить классический тип (СЭД I и СЭД II, 130010). Наибольшая распространенность определена для Республик Татарстан 1:1716 и Карачаево-Черкессия 1:1892. Проводится попытка прогнозирования распространенности Синдрома Элерса – Данло на одной из популяций России на основании значений случайного инбридинга для популяций и случайной изонимии для популяций ранга район.
генетическая эпидемиология
соединительно-тканная дисплазия
синдром элерса-данло
распространенность
1. Амелина С. С., Шокарев Р. А., Кривенцова Н. В., Хлебникова О. В., Ельчинова Г. И., Зинченко Р. А. Генетико-эпидемиологическое изучение Ростовской области // Медицинская генетика. – 2005. – Т. 4, № 8. – С. 371-377.
2. Ельчинова Г. И., Кадошникова М. Ю., Мамедова Р. А. Выявление особенностей генетической структуры популяций с помощью метода описания «генетического ландшафта» // Генетика. – 1991. – Т. 27, № 11. – С. 1994-2001.
3. Ельчинова Г. И., Кривенцова Н. В., Амелина С. С., Тереховская И. Г.,. Валькова Т. И., Вальков Р. А., Зинченко Р. А. Прогнозирование распространенности наследственной патологии на основании инбридинга и случайной изонимии // Медицинская генетика. – 2007. – Т. 6, № 11. – С. 29-33.
4. Зинченко Р. А., Ельчинова Г. И., Галкина В. А., Кириллов А. Г., Абрукова А. В., Петрова Н. В., Тимковская Е. Е., Зинченко С. П., Шокарев Р. А., Морозова А. А., Близнец Е. А., Вассерман Н. Н., Степанова А. А., Поляков А. В., Гинтер Е. К. Дифференциация этнических групп России по генам наследственных болезней // Медицинская генетика. – 2007. – Т.6, № 2. – С. 29-37.
5. Зинченко Р. А., Ельчинова Г. И., Гинтер Е. К. Ассоциация между уровнем индекса эндогамии российских популяций, случайным инбридингом и отягощенностью наследственными болезнями // Медицинская генетика. – 2003. – Т. 2, № 9. – С. 432-436.
6. Зинченко Р. А., Гинтер Е. К. Наследственные болезни в популяциях человека / глава в монографии «Национальное руководство. Наследственные болезни» // под редакцией: Н. П. Бочкова, Е. К. Гинтера, В. П. Пузырева. – М.: ГЭОТАР-Медиа, 2012. – С. 662-704.
7. Кадурина Т. И., Горбунова В. Н. Дисплазия соединительной ткани: руководство для врачей. – СПб.: Элби-Спб, 2009. – 704 с.
8. Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – 3-е изд. – М.: Товарищество научных изданий КМК, Авторская академия, 2007. – 236 с.
9. Мамедова Р. А., Ельчинова Г. И., Старцева Е. А., Козлова С. И., Петрин А. Н., Руденская Г. Е., Хлебникова О. В., Михайлова Л. К., Ефимова М. Н., Щекотихина Ю. А., Прохоров А. Ю., Кадошникова М. Ю., Галкина В. А., Брусинцева О. В., Гинтер Е. К. Медико- и популяционно-генетическое описание Архангельской области // Генетика. – 1996. – Т.32, № 6. – С.837-841.
10. Dong-Dong Wu and Ya-Ping Zhang. Different level of population differentiation among human genes // BMC Evolutionary Biology. – 2011. – Vol.11(16). doi:10.1186/1471-2148-11-16.
11. Online Mendelian Inheritance in Man. URL: https://www.ncbi.nlm.nih.gov/OMIM (дата обращения 28. 02. 2016).
Соединительно-тканные дисплазии (СТД) по современным представлениям – это полиорганная и полисистемная патология с прогредиентным течением, в основе которой лежат дефекты синтеза или катаболизма компонентов внеклеточного матрикса или регуляторов морфогенеза соединительной ткани [7]. Предполагается, что чаще СТД встречаются как недифференцированные дисплазии приобретенного характера, однако большая доля СТД приходится и на моногенные заболевания, чаще с аутосомно-доминантным наследованием. В класс СТД с моногенным характером наследования входят различные формы синдрома Элерса – Данло, несовершенного остеогенеза, нарушений слуха, миопатий, офтальмопатий, кардиомиопатий, дистрофией ногтей и зубов и др.
Для большинства моногенных синдромальных СТД характерны множественные поражения различных органов и систем с преимущественной локализацией патологического процесса в специфической соединительной ткани. Однако для каждого конкретного моногенного заболевания проявления клинической картины зависят в большей степени от типа дефектного коллагена и характера мутационного повреждения гена, включая аллельную и генетическую гетерогенность [7,8].
Одним из основных моногенных заболеваний из группы СТД является синдром Элерса – Данло (СЭД). Заболевание впервые описано в 1892 г. А. Н. Черногубовым, в 1901 г. – E. Ehlers, в 1908 – H. Danlos. Популяционные частоты или распространенность СЭД точно не известна, предполагается 1:5000-1:560000 [7,8]. Минимальными диагностическими критериями заболевания являются: хрупкость и гиперэластичность кожи, гипермобильность суставов, повышенная кровоточивость [7,8]. СЭД представляет группу заболеваний с различными типами, которые выделяют по биохимическим и молекулярным характеристикам, а также на основании клинических проявлений. Классификация СЭД постоянно корректируется.
По данным «Online Mendelian Inheritancein Man» в настоящий момент выделяют 6 основных типов: классический тип (СЭД I и СЭД II, 130010; гены COL1A1, COL5A2, COL5A1); гипермобильный тип – доброкачественная гиперподвижность суставов (СЭД III, 130020 ), сосудистый тип – кожные, суставные изменения и склонность к спонтанным разрывам кишечника и крупных артерий (СЭД IV, 130050; COL3A1), кифосколиотический тип – тяжелая мышечная гипотония и сколиоз при рождении, генерализованная нестабильность суставов, хрупкость склер и глазного яблока ( СЭД VI,225400; PLOD1), «arthrochalasia» тип – низкий рост больных, генерализованная гипермобильность суставов, кровоизлияния и умеренная растяжимость кожи (СЭД VIIA и VIIB, 130060; гены COL1A1, COL1A2) и «dermatosparaxis» тип – «разрыв кожи» (СЭД VIIC, 225410 ; ген ADAMTS2). Шесть ранее выделяемых по классификации Вильфранша (1989) других форм были отнесены к недифференцированным СТД [11].
Сложность диагностики различных типов СЭД, включая недифференцированные формы СТД, обусловлена множественностью единообразных клинических проявлений и выраженный клинический полиморфизм (внутрисемейный, межсемейный и популяционный). Зачастую у отдельных членов одной семьи клинические проявления заболевания варьируют в широких пределах (от легких проявлений до тяжелых органических изменений), что связано как с различной экспрессией гена, так и с функциональными особенностями генов каждого конкретного индивида. Все это значительно затрудняет постановку диагноза конкретных форм Элерса – Данло врачами различных специальностей, т.к. на прием обычно приходит один пациент из семьи с определенным видом поражения соединительной ткани, часто сугубо локальным. В данной ситуации проведение медико-популяционных исследований, дающих возможность одновременно осмотреть всех членов одной семьи в домашних условиях, позволяет учитывать наличие клинического полиморфизма в полной мере и дифференцировать СЭД.
Цель исследования
Целью данного исследования явился анализ распространенности синдрома Элерса – Данло на основании популяционного медико-генетического обследования населения ряда популяций России.
Материалы и методы исследования
Проанализированы результаты генетико-эпидемиологического исследования 10 российских регионов с суммарной численностью обследованного населения более 3,5 млн человек: Кировской (10 районов), Костромской (10 районов), Тверской (2 района), Ростовской (12 районов), Архангельской (5 районов), Брянской (2 района) областей и Краснодарского края (6 районов), Республик Марий Эл (7 районов), Удмуртия (6 районов), Чувашия (6 районов), Башкортостан (8 районов), Татария (8 районов), Адыгея (4 района) и Карачаево-Черкессия (7 районов и г. Черкесск) [1, 4, 6, 9].
Обследование населения проводилось по стандартному протоколу, разработанному в лаборатории генетической эпидемиологии ФГБНУ «МГНЦ» [6]. Частью этого протокола является собственно медико-генетическое обследование, предусматривающее выявление более 2 тыс. моногенных наследственных заболеваний из 4500 известных на настоящий момент. Диагностика НБ осуществлена группой высококвалифицированных специалистов из г. Москвы в полевых условиях (генетиком, неврологом, окулистом, дерматологом, ортопедом). В некоторых случаях для установления диагноза используются молекулярные, биохимические, рентгенологические, электромиографические и др. методы исследований. Далее из материала выделены пациенты с СЭД и проанализирована их распространенность по регионам.
Второй составляющей протокола является изучение генетической структуры изучаемой популяции с использованием методов популяционной статистики (оценки F-статистик Райта, анализа генетических расстояний). Описания генетической структуры изученных популяций, полученные через гены наследственных болезней и через различные популяционные статистики, сравниваются между собой для возможности получения возможных механизмов формирования заболевания и возможности прогноза распространенности заболеваний в необследованных популяциях.
Результаты исследования и их обсуждение
Учитывая, что данное исследование начато более 30 лет назад, когда ДНК-диагностика только начинала свои обороты в РФ, и материал постоянно пополнялся по мере обследования новой популяции, диагноз СЭД регистрировался без деления на типы. Учитывались только семейные случаи заболевания (с 2 и более больными в семье).
Общими клиническими критериями для диагноза являлись: гиперэластичность и повышенная ранимость кожи (с возможным образованием «папирусных» и келоидных рубцов, стрии, возможны кровоизлияния), гипермобильность суставов (наблюдались как врожденные вывихи тазобедренных суставов, так и привычные вывихи локтевых, плечевых, суставов кистей и стоп), патология органа зрения (подвывихи хрусталиков, высокая степень миопии, отслойка сетчатки), изменения скелета (кифозы, сколиозы, деформации грудной клетки, плоскостопие и пр.). Необходимо отметить, что наблюдался выраженный внутрисемейный клинический полиморфизм, выраженность клинических проявлений у отдельных членов семьи существенно варьировала. В большинстве семей клинические проявления различных типов пересекались, однако можно констатировать, что для всех семей можно было предположить классический тип (СЭД I и СЭД II, 130010), связанный с генами COL1A1, COL5A2, COL5A1. В 12 % семей при четкой классической картине заболевания отмечались разрывы сосудов головного мозга и сосудов сердца в молодом возрасте (18–35 лет) у одного из членов родословной. На рис. 1 представлена одна из родословных с СЭД.
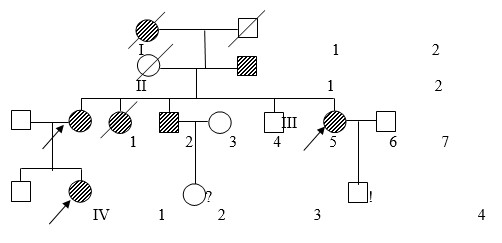
Рис. 1. Родословная семьи К. с СЭД
У большинства членов семьи наблюдались классические проявления синдрома, но выраженность проявлений варьировала. Пробанд IV2 пяти лет имел привычные вывихи в локтевом, плечевом суставах и мелких суставов стоп, гиперэластичную кожу с «папирусными» рубцами на коленях и на голове, продольное и поперечное плоскостопие, деформацию грудной клетки, миопию. У матери III2 дети III7 признаки к моменту осмотра были мягче, но в детстве все вышеперечисленные симптомы ее ребенка отмечались. Дядя пробанда III3 погиб от геморрагического инсульта в 18 лет.
Анализ генетико-эпидемиологических исследований в обследованных нами популяциях европейской части России показал, что распространенность СЭД I и СЭД II составила в среднем 1:9466 с вариацией по популяциям: 1:1716 в Татарстане, 1:1892 в Карачаево-Черкесской Республике, 1:3675 в Брянской области, 1:4169 в Башкортостане, 1:6815 в Ростовской области, 1:14331 в Кировской области, 1:20000 в Архангельской области, 1:29739 в Удмуртии, 1:25333 в Тверской области, 1:30479 в Краснодарском крае, 1:30767 в Марий Эл, 1:37784 в Чувашии, 1:50900 в Адыгее [1, 4, 6, 9]. Как следует из вышеперечисленных данных, наибольшие значения распространенности выявлены в Татарстане – 1:1716 и в Карачаево-Черкесской Республике 1:1892.
Влияние особенностей популяционно-генетической структуры на груз, спектр и территориальной распространение наследственной патологии неоднократно освещалось рядом авторов [2, 5, 6, 10]. Однако работ, касающихся взаимосвязи распространенности отдельных нозологических форм с параметрами популяционно-генетической структуры, очень мало.
При медико-генетическом обследовании Ростовской области была представлена принципиальная возможность прогнозирования распространенности наследственной патологии через параметры популяционной структуры [3]. Было показано, что наиболее устойчивыми во времени являются оценки параметров популяционной структуры, полученные для популяций ранга «район». Получив высокие корреляции между основными параметрами популяционно-генетической структуры и распространенностью наследственной патологии [3], мы не стали проверять еще раз принципиальную возможность подобного прогнозирования, а попробовали применить ту же самую методику для отдельных нозологий, в частности СЭД.
Была выбрана популяция – Республика Татарстан, в которой медико-генетическое обследование было проведено в 8 районах, а генетическая структура через оценку параметров случайной изонимии Барраи оценена в 16 районах. Из всего спектра выявленных наследственных заболеваний в Татарстане (РТ) мы выбрали наиболее распространенную АД-наследуемую патологию, которой оказался синдром Элерса – Данло – 154 больных, распространенность составила 1:1716.
В табл. 1 представлены значения случайной изонимии Барраи, число больных и распространенность СЭД в 8 районах Татарстана.
Таблица 1
Наблюдаемые значения больных СЭД и случайная изонимия Барраи для популяций ранга «район» в Татарстане
район | численность населения | число больных | распространенность | случайная изонимия |
Арский | 51607 | 25 | 0,000484 | 0,003536 |
Атнинский | 13800 | 21 | 0,001522 | 0,006519 |
Кукморский | 47414 | 16 | 0,000337 | 0,002878 |
Дрожжановский | 25841 | 20 | 0,000774 | 0,002476 |
Буинский | 45144 | 21 | 0,000465 | 0,002308 |
Актанышский | 31790 | 22 | 0,000692 | 0,003256 |
Муслюмовский | 19638 | 21 | 0,001069 | 0,002987 |
Мензелинский | 29076 | 8 | 0,000275 | 0,001692 |
Коэффициент корреляции распространенности синдрома Элерса – Данло и случайной изонимии Барраи для популяций ранга «район» составил 0,82±0,24, что свидетельствует о значительном влиянии инбридинга на распространенность этого заболевания (рис. 2).
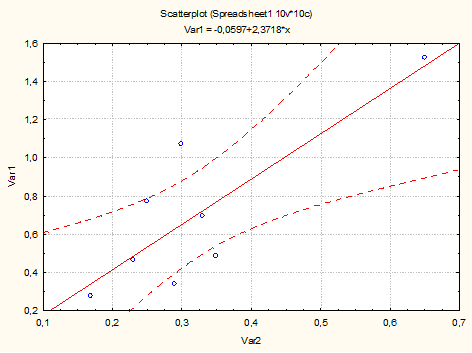
Рис.2. Зависимость распространенности СЭД от случайной изонимии
Случайная изонимия Барраи отличается от случайного инбридинга Райта лишь коэффициентом (что не влияет на коэффициент корреляции), но имеет более высокие абсолютные значения, поэтому является более предпочтительной при моделировании. Расчеты выполнены без учета этнической принадлежности пациентов. Это позволило нам сделать прогноз распространенности СЭД для еще 8 необследованных районов, для которых были известны значения параметров Барраи. Значения прогноза представлены в табл. 2.
Таблица 2
Случайная изонимия Барраи и прогноз распространенности СЭД и числа больных для необследованных районов Татарстана
район | случайная изонимия | численность населения (тыс. чел.) | рассчитанная распространенность | прогнозируемое число больных |
Ютазинский | 0,002581 | 23,2 | 0,000552 | 13 |
Пестречинский | 0,001699 | 28,6 | 0,000343 | 10 |
Балтасинский | 0,003887 | 34,3 | 0,000862 | 30 |
Алькеевский | 0,003030 | 21,0 | 0,000659 | 14 |
Бугульминский | 0,000840 | 112,3 | 0,00014 | 16 |
Нурлатский | 0,001645 | 60,4 | 0,00033 | 20 |
Черемшанский | 0,002345 | 21,1 | 0,000496 | 10 |
Азнакаевский | 0,002834 | 64,3 | 0,000612 | 39 |
Заключение
На основании генетико-эпидемиологических исследований проанализирована распространенность СЭД в ряде популяций России: Кировской, Костромской, Тверской, Ростовской, Архангельской, Брянской областей и Краснодарского края, Республик Марий Эл, Удмуртия, Чувашия, Башкортостан, Татария, Адыгея и Карачаево-Черкессия. Во всех семьях выявлен классический тип (СЭД I и СЭД II, 130010). Накопление СЭД выявлено в Республике Татарстан (1:1716) и Карачаево-Черкессии (1:1892). На основании имеющихся данных о распространенности СЭД в 8 районах Татарстана и значений случайного инбридинга проведено прогнозирование значений распространенности в необследованных районах РТ.
Работа выполнена при частичном финансировании РФФИ (14-04-00525, 15-04-01859).
Библиографическая ссылка
Макаов А.Х., Ельчинова Г.И., Галкина В.А., Куцев С.И., Зинченко Р.А. РАСПРОСТРАНЕННОСТЬ СИНДРОМА ЭЛЕРСА-ДАНЛО В РЯДЕ ПОПУЛЯЦИЙ РОССИИ // Современные проблемы науки и образования. – 2016. – № 3.;
URL: https://science-education.ru/ru/article/view?id=24395 (дата обращения: 14.10.2020).
Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)
Источник