
Синдром элерса данлоса частота встречаемости

- Авторы
- Резюме
- Файлы
- Ключевые слова
- Литература
Макаов А.Х.
1
Ельчинова Г.И.
2
Галкина В.А.
2
Куцев С.И.
2, 3
Зинченко Р.А.
2, 3
1 Муниципальное бюджетное лечебно-профилактическое учреждение «Хабезская центральная районная больница»
2 Федеральное государственное бюджетное научное учреждение «Медико-генетический научный центр»
3 ГБОУ ВПО Российский национальный исследовательский медицинский университет имени Н.И.Пирогова»
Целью данного исследования явился анализ распространенности синдрома Элерса – Данло (СЭД ) на основании популяционного медико-генетического обследования населения ряда популяций России. Проведен анализ распространенности синдрома СЭД в ряде популяций европейской части России: Кировской, Костромской, Тверской, Ростовской, Архангельской, Брянской областей и Краснодарского края, Республик Марий Эл, Удмуртия, Чувашия, Башкортостан, Татария, Адыгея и Карачаево-Черкессия. В большинстве семей клинические проявления различных типов СЭД пересекались, однако можно констатировать, что для всех семей можно было предположить классический тип (СЭД I и СЭД II, 130010). Наибольшая распространенность определена для Республик Татарстан 1:1716 и Карачаево-Черкессия 1:1892. Проводится попытка прогнозирования распространенности Синдрома Элерса – Данло на одной из популяций России на основании значений случайного инбридинга для популяций и случайной изонимии для популяций ранга район.
генетическая эпидемиология
соединительно-тканная дисплазия
синдром элерса-данло
распространенность
1. Амелина С. С., Шокарев Р. А., Кривенцова Н. В., Хлебникова О. В., Ельчинова Г. И., Зинченко Р. А. Генетико-эпидемиологическое изучение Ростовской области // Медицинская генетика. – 2005. – Т. 4, № 8. – С. 371-377.
2. Ельчинова Г. И., Кадошникова М. Ю., Мамедова Р. А. Выявление особенностей генетической структуры популяций с помощью метода описания «генетического ландшафта» // Генетика. – 1991. – Т. 27, № 11. – С. 1994-2001.
3. Ельчинова Г. И., Кривенцова Н. В., Амелина С. С., Тереховская И. Г.,. Валькова Т. И., Вальков Р. А., Зинченко Р. А. Прогнозирование распространенности наследственной патологии на основании инбридинга и случайной изонимии // Медицинская генетика. – 2007. – Т. 6, № 11. – С. 29-33.
4. Зинченко Р. А., Ельчинова Г. И., Галкина В. А., Кириллов А. Г., Абрукова А. В., Петрова Н. В., Тимковская Е. Е., Зинченко С. П., Шокарев Р. А., Морозова А. А., Близнец Е. А., Вассерман Н. Н., Степанова А. А., Поляков А. В., Гинтер Е. К. Дифференциация этнических групп России по генам наследственных болезней // Медицинская генетика. – 2007. – Т.6, № 2. – С. 29-37.
5. Зинченко Р. А., Ельчинова Г. И., Гинтер Е. К. Ассоциация между уровнем индекса эндогамии российских популяций, случайным инбридингом и отягощенностью наследственными болезнями // Медицинская генетика. – 2003. – Т. 2, № 9. – С. 432-436.
6. Зинченко Р. А., Гинтер Е. К. Наследственные болезни в популяциях человека / глава в монографии «Национальное руководство. Наследственные болезни» // под редакцией: Н. П. Бочкова, Е. К. Гинтера, В. П. Пузырева. – М.: ГЭОТАР-Медиа, 2012. – С. 662-704.
7. Кадурина Т. И., Горбунова В. Н. Дисплазия соединительной ткани: руководство для врачей. – СПб.: Элби-Спб, 2009. – 704 с.
8. Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – 3-е изд. – М.: Товарищество научных изданий КМК, Авторская академия, 2007. – 236 с.
9. Мамедова Р. А., Ельчинова Г. И., Старцева Е. А., Козлова С. И., Петрин А. Н., Руденская Г. Е., Хлебникова О. В., Михайлова Л. К., Ефимова М. Н., Щекотихина Ю. А., Прохоров А. Ю., Кадошникова М. Ю., Галкина В. А., Брусинцева О. В., Гинтер Е. К. Медико- и популяционно-генетическое описание Архангельской области // Генетика. – 1996. – Т.32, № 6. – С.837-841.
10. Dong-Dong Wu and Ya-Ping Zhang. Different level of population differentiation among human genes // BMC Evolutionary Biology. – 2011. – Vol.11(16). doi:10.1186/1471-2148-11-16.
11. Online Mendelian Inheritance in Man. URL: https://www.ncbi.nlm.nih.gov/OMIM (дата обращения 28. 02. 2016).
Соединительно-тканные дисплазии (СТД) по современным представлениям – это полиорганная и полисистемная патология с прогредиентным течением, в основе которой лежат дефекты синтеза или катаболизма компонентов внеклеточного матрикса или регуляторов морфогенеза соединительной ткани [7]. Предполагается, что чаще СТД встречаются как недифференцированные дисплазии приобретенного характера, однако большая доля СТД приходится и на моногенные заболевания, чаще с аутосомно-доминантным наследованием. В класс СТД с моногенным характером наследования входят различные формы синдрома Элерса – Данло, несовершенного остеогенеза, нарушений слуха, миопатий, офтальмопатий, кардиомиопатий, дистрофией ногтей и зубов и др.
Для большинства моногенных синдромальных СТД характерны множественные поражения различных органов и систем с преимущественной локализацией патологического процесса в специфической соединительной ткани. Однако для каждого конкретного моногенного заболевания проявления клинической картины зависят в большей степени от типа дефектного коллагена и характера мутационного повреждения гена, включая аллельную и генетическую гетерогенность [7,8].
Одним из основных моногенных заболеваний из группы СТД является синдром Элерса – Данло (СЭД). Заболевание впервые описано в 1892 г. А. Н. Черногубовым, в 1901 г. – E. Ehlers, в 1908 – H. Danlos. Популяционные частоты или распространенность СЭД точно не известна, предполагается 1:5000-1:560000 [7,8]. Минимальными диагностическими критериями заболевания являются: хрупкость и гиперэластичность кожи, гипермобильность суставов, повышенная кровоточивость [7,8]. СЭД представляет группу заболеваний с различными типами, которые выделяют по биохимическим и молекулярным характеристикам, а также на основании клинических проявлений. Классификация СЭД постоянно корректируется.
По данным «Online Mendelian Inheritancein Man» в настоящий момент выделяют 6 основных типов: классический тип (СЭД I и СЭД II, 130010; гены COL1A1, COL5A2, COL5A1); гипермобильный тип – доброкачественная гиперподвижность суставов (СЭД III, 130020 ), сосудистый тип – кожные, суставные изменения и склонность к спонтанным разрывам кишечника и крупных артерий (СЭД IV, 130050; COL3A1), кифосколиотический тип – тяжелая мышечная гипотония и сколиоз при рождении, генерализованная нестабильность суставов, хрупкость склер и глазного яблока ( СЭД VI,225400; PLOD1), «arthrochalasia» тип – низкий рост больных, генерализованная гипермобильность суставов, кровоизлияния и умеренная растяжимость кожи (СЭД VIIA и VIIB, 130060; гены COL1A1, COL1A2) и «dermatosparaxis» тип – «разрыв кожи» (СЭД VIIC, 225410 ; ген ADAMTS2). Шесть ранее выделяемых по классификации Вильфранша (1989) других форм были отнесены к недифференцированным СТД [11].
Сложность диагностики различных типов СЭД, включая недифференцированные формы СТД, обусловлена множественностью единообразных клинических проявлений и выраженный клинический полиморфизм (внутрисемейный, межсемейный и популяционный). Зачастую у отдельных членов одной семьи клинические проявления заболевания варьируют в широких пределах (от легких проявлений до тяжелых органических изменений), что связано как с различной экспрессией гена, так и с функциональными особенностями генов каждого конкретного индивида. Все это значительно затрудняет постановку диагноза конкретных форм Элерса – Данло врачами различных специальностей, т.к. на прием обычно приходит один пациент из семьи с определенным видом поражения соединительной ткани, часто сугубо локальным. В данной ситуации проведение медико-популяционных исследований, дающих возможность одновременно осмотреть всех членов одной семьи в домашних условиях, позволяет учитывать наличие клинического полиморфизма в полной мере и дифференцировать СЭД.
Цель исследования
Целью данного исследования явился анализ распространенности синдрома Элерса – Данло на основании популяционного медико-генетического обследования населения ряда популяций России.
Материалы и методы исследования
Проанализированы результаты генетико-эпидемиологического исследования 10 российских регионов с суммарной численностью обследованного населения более 3,5 млн человек: Кировской (10 районов), Костромской (10 районов), Тверской (2 района), Ростовской (12 районов), Архангельской (5 районов), Брянской (2 района) областей и Краснодарского края (6 районов), Республик Марий Эл (7 районов), Удмуртия (6 районов), Чувашия (6 районов), Башкортостан (8 районов), Татария (8 районов), Адыгея (4 района) и Карачаево-Черкессия (7 районов и г. Черкесск) [1, 4, 6, 9].
Обследование населения проводилось по стандартному протоколу, разработанному в лаборатории генетической эпидемиологии ФГБНУ «МГНЦ» [6]. Частью этого протокола является собственно медико-генетическое обследование, предусматривающее выявление более 2 тыс. моногенных наследственных заболеваний из 4500 известных на настоящий момент. Диагностика НБ осуществлена группой высококвалифицированных специалистов из г. Москвы в полевых условиях (генетиком, неврологом, окулистом, дерматологом, ортопедом). В некоторых случаях для установления диагноза используются молекулярные, биохимические, рентгенологические, электромиографические и др. методы исследований. Далее из материала выделены пациенты с СЭД и проанализирована их распространенность по регионам.
Второй составляющей протокола является изучение генетической структуры изучаемой популяции с использованием методов популяционной статистики (оценки F-статистик Райта, анализа генетических расстояний). Описания генетической структуры изученных популяций, полученные через гены наследственных болезней и через различные популяционные статистики, сравниваются между собой для возможности получения возможных механизмов формирования заболевания и возможности прогноза распространенности заболеваний в необследованных популяциях.
Результаты исследования и их обсуждение
Учитывая, что данное исследование начато более 30 лет назад, когда ДНК-диагностика только начинала свои обороты в РФ, и материал постоянно пополнялся по мере обследования новой популяции, диагноз СЭД регистрировался без деления на типы. Учитывались только семейные случаи заболевания (с 2 и более больными в семье).
Общими клиническими критериями для диагноза являлись: гиперэластичность и повышенная ранимость кожи (с возможным образованием «папирусных» и келоидных рубцов, стрии, возможны кровоизлияния), гипермобильность суставов (наблюдались как врожденные вывихи тазобедренных суставов, так и привычные вывихи локтевых, плечевых, суставов кистей и стоп), патология органа зрения (подвывихи хрусталиков, высокая степень миопии, отслойка сетчатки), изменения скелета (кифозы, сколиозы, деформации грудной клетки, плоскостопие и пр.). Необходимо отметить, что наблюдался выраженный внутрисемейный клинический полиморфизм, выраженность клинических проявлений у отдельных членов семьи существенно варьировала. В большинстве семей клинические проявления различных типов пересекались, однако можно констатировать, что для всех семей можно было предположить классический тип (СЭД I и СЭД II, 130010), связанный с генами COL1A1, COL5A2, COL5A1. В 12 % семей при четкой классической картине заболевания отмечались разрывы сосудов головного мозга и сосудов сердца в молодом возрасте (18–35 лет) у одного из членов родословной. На рис. 1 представлена одна из родословных с СЭД.
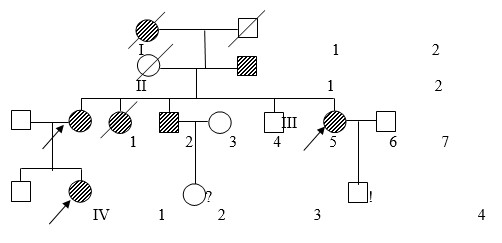
Рис. 1. Родословная семьи К. с СЭД
У большинства членов семьи наблюдались классические проявления синдрома, но выраженность проявлений варьировала. Пробанд IV2 пяти лет имел привычные вывихи в локтевом, плечевом суставах и мелких суставов стоп, гиперэластичную кожу с «папирусными» рубцами на коленях и на голове, продольное и поперечное плоскостопие, деформацию грудной клетки, миопию. У матери III2 дети III7 признаки к моменту осмотра были мягче, но в детстве все вышеперечисленные симптомы ее ребенка отмечались. Дядя пробанда III3 погиб от геморрагического инсульта в 18 лет.
Анализ генетико-эпидемиологических исследований в обследованных нами популяциях европейской части России показал, что распространенность СЭД I и СЭД II составила в среднем 1:9466 с вариацией по популяциям: 1:1716 в Татарстане, 1:1892 в Карачаево-Черкесской Республике, 1:3675 в Брянской области, 1:4169 в Башкортостане, 1:6815 в Ростовской области, 1:14331 в Кировской области, 1:20000 в Архангельской области, 1:29739 в Удмуртии, 1:25333 в Тверской области, 1:30479 в Краснодарском крае, 1:30767 в Марий Эл, 1:37784 в Чувашии, 1:50900 в Адыгее [1, 4, 6, 9]. Как следует из вышеперечисленных данных, наибольшие значения распространенности выявлены в Татарстане – 1:1716 и в Карачаево-Черкесской Республике 1:1892.
Влияние особенностей популяционно-генетической структуры на груз, спектр и территориальной распространение наследственной патологии неоднократно освещалось рядом авторов [2, 5, 6, 10]. Однако работ, касающихся взаимосвязи распространенности отдельных нозологических форм с параметрами популяционно-генетической структуры, очень мало.
При медико-генетическом обследовании Ростовской области была представлена принципиальная возможность прогнозирования распространенности наследственной патологии через параметры популяционной структуры [3]. Было показано, что наиболее устойчивыми во времени являются оценки параметров популяционной структуры, полученные для популяций ранга «район». Получив высокие корреляции между основными параметрами популяционно-генетической структуры и распространенностью наследственной патологии [3], мы не стали проверять еще раз принципиальную возможность подобного прогнозирования, а попробовали применить ту же самую методику для отдельных нозологий, в частности СЭД.
Была выбрана популяция – Республика Татарстан, в которой медико-генетическое обследование было проведено в 8 районах, а генетическая структура через оценку параметров случайной изонимии Барраи оценена в 16 районах. Из всего спектра выявленных наследственных заболеваний в Татарстане (РТ) мы выбрали наиболее распространенную АД-наследуемую патологию, которой оказался синдром Элерса – Данло – 154 больных, распространенность составила 1:1716.
В табл. 1 представлены значения случайной изонимии Барраи, число больных и распространенность СЭД в 8 районах Татарстана.
Таблица 1
Наблюдаемые значения больных СЭД и случайная изонимия Барраи для популяций ранга «район» в Татарстане
район | численность населения | число больных | распространенность | случайная изонимия |
Арский | 51607 | 25 | 0,000484 | 0,003536 |
Атнинский | 13800 | 21 | 0,001522 | 0,006519 |
Кукморский | 47414 | 16 | 0,000337 | 0,002878 |
Дрожжановский | 25841 | 20 | 0,000774 | 0,002476 |
Буинский | 45144 | 21 | 0,000465 | 0,002308 |
Актанышский | 31790 | 22 | 0,000692 | 0,003256 |
Муслюмовский | 19638 | 21 | 0,001069 | 0,002987 |
Мензелинский | 29076 | 8 | 0,000275 | 0,001692 |
Коэффициент корреляции распространенности синдрома Элерса – Данло и случайной изонимии Барраи для популяций ранга «район» составил 0,82±0,24, что свидетельствует о значительном влиянии инбридинга на распространенность этого заболевания (рис. 2).
Рис.2. Зависимость распространенности СЭД от случайной изонимии
Случайная изонимия Барраи отличается от случайного инбридинга Райта лишь коэффициентом (что не влияет на коэффициент корреляции), но имеет более высокие абсолютные значения, поэтому является более предпочтительной при моделировании. Расчеты выполнены без учета этнической принадлежности пациентов. Это позволило нам сделать прогноз распространенности СЭД для еще 8 необследованных районов, для которых были известны значения параметров Барраи. Значения прогноза представлены в табл. 2.
Таблица 2
Случайная изонимия Барраи и прогноз распространенности СЭД и числа больных для необследованных районов Татарстана
район | случайная изонимия | численность населения (тыс. чел.) | рассчитанная распространенность | прогнозируемое число больных |
Ютазинский | 0,002581 | 23,2 | 0,000552 | 13 |
Пестречинский | 0,001699 | 28,6 | 0,000343 | 10 |
Балтасинский | 0,003887 | 34,3 | 0,000862 | 30 |
Алькеевский | 0,003030 | 21,0 | 0,000659 | 14 |
Бугульминский | 0,000840 | 112,3 | 0,00014 | 16 |
Нурлатский | 0,001645 | 60,4 | 0,00033 | 20 |
Черемшанский | 0,002345 | 21,1 | 0,000496 | 10 |
Азнакаевский | 0,002834 | 64,3 | 0,000612 | 39 |
Заключение
На основании генетико-эпидемиологических исследований проанализирована распространенность СЭД в ряде популяций России: Кировской, Костромской, Тверской, Ростовской, Архангельской, Брянской областей и Краснодарского края, Республик Марий Эл, Удмуртия, Чувашия, Башкортостан, Татария, Адыгея и Карачаево-Черкессия. Во всех семьях выявлен классический тип (СЭД I и СЭД II, 130010). Накопление СЭД выявлено в Республике Татарстан (1:1716) и Карачаево-Черкессии (1:1892). На основании имеющихся данных о распространенности СЭД в 8 районах Татарстана и значений случайного инбридинга проведено прогнозирование значений распространенности в необследованных районах РТ.
Работа выполнена при частичном финансировании РФФИ (14-04-00525, 15-04-01859).
Библиографическая ссылка
Макаов А.Х., Ельчинова Г.И., Галкина В.А., Куцев С.И., Зинченко Р.А. РАСПРОСТРАНЕННОСТЬ СИНДРОМА ЭЛЕРСА-ДАНЛО В РЯДЕ ПОПУЛЯЦИЙ РОССИИ // Современные проблемы науки и образования. – 2016. – № 3.;
URL: https://science-education.ru/ru/article/view?id=24395 (дата обращения: 13.01.2021).

Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)
Источник
Клинические характеристики
Гипермобильный тип синдрома Элерса-Данло — это заболевание соединительной ткани, которое сопровождается выраженной гипермобильностью суставов и хронической болью в суставах, связках, сухожилиях, позвоночнике.
Частота встречаемости: 1 на 10,000-15,000 человек.
Гипермобильный тип СЭД считается самым легким типом, хотя он может давать довольно серьезные осложнения. Главным образом это касается опорно-двигательного аппарата. При этом типе очень часто происходят вывихи и подвывихи в различных суставах, они могут возникать спонтанно или после минимальной травмы. Обычно они очень болезненны. Довольно рано у этих пациентов развиваются дегенеративные заболевания суставов и позвоночника. Этот тип сопровождает хроническая боль, которая может быть не связана напрямую с вывихами, и это может серьезно влиять на качество жизни.
Кожа при гипермобильном типе СЭД может быть мягкой, бархатистой и чрезмерно растяжимой, но не всегда. Довольно часто встречается легкое появление синяков. Часты функциональные расстройства желудочно-кишечного тракта, а также дисфункция вегетативной нервной системы. Может присутствовать расширение корня аорты, но обычно в слабой степени и без повышенного риска расслоения и разрыва.
Постановка диагноза
Диагностика гипермобильного типа СЭД основывается полностью на результатах клинического осмотра и семейной истории. Генетическая мутация, связанная с гипермобильным типом СЭД, пока не найдена.
Диагностические критерии для него были приняты в 1997 году. Комбинация двух основных критериев имеет высокую специфичность для гипермобильного типа СЭД. Наличие одного или более дополнительных критериев дополняет картину, но не является достаточным для диагноза.
Основные критерии:
- Генерализованная гипермобильность суставов (5 и больше баллов по шкале Бейтона). Однако в некоторых случаях люди с гипермобильным типом могут набирать меньшее количество баллов при условии, что у них есть объективно сильно выраженная гибкость суставов. Также гипермобильность может сильно варьироваться в зависимости от пола, возраста и этнической принадлежности.
- Проявления со стороны кожи (чрезмерно растяжимая и/или мягкая, бархатистая кожа). Растяжимость кожи должна измеряться в нейтральном месте, например, участок руки от запястья до локтя. Необходимо оттянуть кожу, пока не почувствуется сопротивление. У маленьких детей очень сложно измерить растяжимость кожи в связи с обилием подкожного жира. Чрезмерная хрупкость кожи, атрофические шрамы и плохое заживление ссадин нехарактерны для гипермобильного типа.
Дополнительные критерии:
- Повторяющиеся вывихи и подвывихи суставов.
- Хроническая боль в суставах, связках, мышцах.
- Гипермобильность суставов у родственников (семейная история).
- Легкое появление синяков.
- Функциональные расстройства пищеварения (функциональный гастрит, синдром раздраженного кишечника).
- Высокое и узкое небо.
- Скученность зубов.
- Постуральная ортостатическая тахикардия.
У пациентов с гипермобильным типом СЭД также могут быть следующие симптомы:
- тендениты, бурситы и остеоартрит могут развиваться как следствие гипермобильности суставов;
- пониженная плотность костей даже в раннем возрасте;
- проблемы со сном;
- хроническая усталость;
- головные боли, включая мигрень;
- расстройства вегетативной нервной системы;
- пролапс митрального клапана;
- дисфункция височно-нижнечелюстных суставов;
- чувствительные десна, склонные к кровоточивости;
- миопия;
- пониженная чувствительность к местной анестезии.
Действия после постановки диагноза
Сразу после того, как человеку был поставлен диагноз “гипермобильный тип синдрома Элерса-Данло”, необходимо оценить масштаб заболевания при помощи следующих действий:
- клинический осмотр пациента, выявление жалоб со стороны опорно-двигательного аппарата, сердечно-сосудистой и пищеварительной систем и стоматологический осмотр;
- эхокардиограмма (УЗИ сердца) с измерением диаметра аорты;
- для пациентов с постуральной ортостатической тахикардией (увеличение частоты пульса после смены положения тела из горизонтального в вертикальное) необходимо провести тилт-тест;
- для пациентов со значительным уровнем боли и хронической усталостью можно провести обследование на предмет выявления возможных причин: дефицит витаминов D и B12, дефицит железа и фолиевой кислоты, целиакия. Эти расстройства могут быть связаны с проблемами с пищеварением у пациентов с гипермобильным типом.
- для диагностики остеопении необходимо сделать денситометрию.
Поскольку СЭД — это наследственное заболевание, после постановки диагноза очень важно провести осмотр родственников больного первой степени родства (родителей и детей). Если СЭД обнаруживается у родителей больного, необходимо также обследование родных братьев и сестер.
Терапия симптомов
Людям с гипермобильным типом СЭД необходимо укреплять мышцы, чтобы стабилизировать суставы. Предпочтение стоит отдавать упражнениям без веса и лечебной физкультуре, а также плаванию. Особенно это важно для детей с гипотонией и замедленным моторным развитием.
Возможно, для облегчения боли необходимо снизить физическую нагрузку, изменить свой образ жизни и профессию. В преодолении трудностей, связанных с этими ограничениями, может помочь психотерапия.
Для временного уменьшения боли может помочь массаж, физиотерапия и акупунктура.
Уменьшить боли в спине и шее может тщательно подобранные матрас и подушка.
Врач должен серьезно подойти к решению вопроса снятия боли у пациента с гипермобильным типом СЭД, подобрав соответствующую медикаментозную терапию.
Профилактика
Чтобы стабилизировать больные суставы, используют ортезы и бандажи. Трудности могут возникать только с поддержкой плечевых и тазобедренных суставов. Для стабилизации суставов пальцев рук используют специальные кольца. Для подбора оптимальных ортопедических средств желательно проконсультироваться с врачом-ортопедом.
При сильной нестабильности и болях в суставах ног следует задуматься об использовании инвалидного кресла.
Если имеется расширение аорты и/или пролапс митрального клапана, необходимо каждый год делать эхокардиограмму (УЗИ сердца).
При пониженной плотности костей рекомендуется принимать кальций и витамин D.
Следует избегать: видов спорта, которые перегружают суставы (контактных видов спорта, боевых искусств, футбола, бега и т.д.), упражнений для растяжки, слишком тяжелых упражнений и упражнений с весом, мануальной терапии в связи с повышенным риском получить травму.
Беременность
У женщин с гипермобильным типом синдрома Элерса-Данло роды могут проходить очень стремительно. Естественные роды могут привести к вывиху тазобедренных суставов, но кесарево сечение несет такие же риски, как для здорового человека.
Во время беременности увеличивается гибкость суставов и, соответственно, боль в суставах, особенно в третьем триместре. Вероятность вывихов и подвывихов также возрастает.
Если имеется расширение аорты, необходимо делать УЗИ сердца каждый триместр.
Наследование
Гипермобильный тип синдрома Элерса-Данло наследуется аутосомно-доминантно. Это значит, шанс передачи этого заболевания потомкам — 50%. Также примерно 50% больных унаследовали его от родителя, а у других 50% оно возникло спонтанно.
Информация взята из GeneReviews.
Источник