
Синдром эллиса ван кревельда фото

Ричард Эллис из Эдинбурга и Симон ван Кревелд из Амстердама, впервые описали синдром Эллиса-ван Кревелда. Они встретились в купе поезда во время поездки на конференцию по педиатрии в Англии в конце 1930-х годов и обнаружили, что в их докладах описывалось два пациента с одинаковыми проявлениями, что и заставило их думать, что у их пациентов один синдром. В 1940 году Эллис и ван Кревелд формально описали этот синдром, который в будущем будет нести в своем названии их имена, хотя изначально, они назвали его как хондроэктодермальная дисплазия. Непропорциональная карликовость, полидактилия, эктодермальная дисплазия и высокая частота развития врожденных пороков сердца, характеризуют этот синдром.
Синдром Эллиса-ван Кревелда. Эпидемиология
Синдром Эллиса-ван Кревелда развивается у 1 на 60000 живорожденных.
Синдром Эллиса-ван Кревелда. Причины
Синдром Эллиса-ван Кревелда имеет аутосомно-рецессивный тип наследования. Мутации в гене EVC, который находится на полосе 4p16, несут ответственность за развитие этого синдрома. Кодируемый этим геном белок состоит из 992 аминокислоты. Мутации в гене EVC1 часто встречаются у амишей и бразильцев. А в последнее время, были еще найдены мутации во втором гене, EVC2, которые впервые были выявлены и описаны у ребенка с синдромом Эллиса-ван Кревелда. В настоящее время, синдром Эллиса-ван Кревелда считается цилиопатией. Каркасные условия, относящиеся к группе цилиопатий, включают в себя (1) реберную полидактилию, в том числе удушающую грудную дистрофию (синдром Жена) и синдром Эллиса-ван Кревелда, (2) синдром Сенсенбреннера и (3) дизостоз Вейерса.
Синдром Эллиса-ван Кревелда. Фото
Новорожденный с синдромом Эллиса-ван Кревелда. Обратите внимание на узкую грудь.
Зубы у новорожденного
Полидактилия
Новорожденный с синдромом Эллиса-ван Кревелда. Обратите внимание на узкую грудь и на непропорциональную карликовость.
Синдром Эллиса-ван Кревелда. Патофизиология
Патофизиология неизвестна. Однако, недавняя идентификация гена EVC должна привести к лучшему пониманию этой болезни.
Синдром Эллиса-ван Кревелда. Симптомы и проявления
- В предродовой период, внутриутробная задержка роста, скелетные аномалии и пороки сердца могут быть обнаружены на УЗИ.
- Неонатальные проявления могут включать в себя небольшой размер при рождении, медленный рост и скелетные аномалии. В некоторых случаях у ребенка в этом периоде уже могут виднеться зубы.
- Болезнь сердца может проявляться в виде проблем в росте, цианозе, одышке или в виде других признаков, указывающих на сердечную недостаточность.
- Большинство пациентов имеют нормальный интеллект. Но иногда, дети могут иметь пороки развития головного мозга и задержки в развитии.
Физическое обследование
Клиническая тетрада синдрома Эллиса-ван Кревелда состоит из хондродистрофии, полидактилии, эктодермальной дисплазии и сердечных аномалий.
Хондродистрофия (наиболее общая черта, затрагивающая трубчатые кости)
- Непропорциональная карликовость (маленький рост, средняя высота взрослого человека составляет 109-155 см)
- Прогрессивное укорочение конечностей, симметрично затрагивающее предплечья и голени
Полидактилия
- Двусторонняя
- Полидактилия наблюдается на руках в большинстве случаев, на ногах встречается в 10% случаев
Эктодермальная дисплазия (наблюдается у 93% пациентов)
- Дистрофические и рыхлые ногти. В некоторых случаях ногти могут полностью отсутствовать.
- У некоторых детей уже в неонатальном возрасте могут присутствовать зубы, частичная анодонтия. Гипоплазия эмали может привести к развитию аномальной формы зубов.
Врожденные пороки сердца
- Пороки сердца встречаются у 50-60% больных. Наиболее распространенные аномалии встречаются в предсердиях.
- Сердечные аномалии являются основной причиной короткой продолжительности жизни.
Другие аномалии также могут присутствовать.
Аномалии опорно-двигательного аппарата включают узкую грудную клетку, которая часто приводит к затрудненному дыханию, потрескивание в коленных суставах, поясничный лордоз, широкие руки и ноги, а пальцы в форме колбасок. Аномалии ротовой полости включают:
- Верхняя губа может быть присоединена к десне верхней челюсти.
- Верхняя губа может быть короткой.
- Зубы могут проявляться уже при рождении.
Аномалии мочеполовой системы включают гипоспадии, гипоплазии, крипторхизм, атрезию вульвы, фокусное расширение почечных канальцев в медуллярной области, нефрокальциноз, почечную агенезию.
Синдром Эллиса-ван Кревелда. Диагностика
- Секвенированием генов EVC и EVC2 можно определить мутации у двух трети пациентов с синдромом Эллиса-ван Кревелда.
- Рентгенография грудной клетки, ЭКГ, эхокардиография.
- МРТ мозга.
- УЗИ почек.
- Обследование глаз.
Синдром Эллиса-ван Кревелда. Лечение
Лечение синдрома Эллиса-ван Кревелда является междисциплинарным. У всех пациентов должен проводиться контроль дыхательной недостаточности, рецидивирующих инфекций дыхательных путей и сердечной недостаточности.
Стоматологическая помощь:
- Аномальные зубы у новорожденных должны быть удалены, поскольку они могут нарушить рост нормальных зубов.
- Должна проводиться профилактика кариеса, которая включает в себя составление диеты и обучение гигиене.
- Частичные съемные протезы могут улучшить жевание, эстетику и речь у детей.
У взрослых лиц, стоматологи должны применить имплантаты и протезы для замены врожденно недостающих зубов. Низкий рост возможно откорректировать гормоном роста, но только если пациент будет иметь дефицит гормона роста.
Хирургическое вмешательство
Некоторые ортопедические процедуры могут быть полезными в решении вопросов связанных с полидактилией и с другими ортопедическими аномалиями. В некоторых случаях может потребоваться операция на сердце. Но к этому подходу необходимо подходить с большой осторожностью, так как хирургическое исправление врожденных пороков сердца, при синдроме Эллиса-ван Кревелда, связано с существенной послеоперационной смертностью. В частности, у некоторых детей после операции на сердце могут развиться проблемы с дыхательной системой, что имеет центральное значение по отношению к послеоперационному исходу.
Помимо аномалий в легких, заячья губа и волчья пасть также могут усугубить послеоперационный прогноз для пациента. Аномалии в гайморовых пазухах или деформации нижней челюсти, также могут привести к трудностям в установке маски аппарата искусственной вентиляции легких и масок от других жизненно важных для пациента аппаратов. Эти проблемы обязательно должны быть определены в ходе предоперационного обследования.
У некоторых детей с сжатой грудной клеткой, хирурги могут расширить ее. Стоматологическая помощь, как правило, также необходима.
Синдром Эллиса-ван Кревелда. Прогноз
- Около 50% пациентов с синдромом Эллиса-ван Кревельда умирают в раннем детстве, в следствие кардиореспираторных проблем. Большинство оставшихся в живых имеют нормальный интеллект. Грудная дисплазия, которая приводит к дыхательной недостаточности и сердечные аномалии являются самыми распространенными причинами ранней смерти. Дети, которым удастся пережить младенчество и у которых будут устранены все самые угрожающие жизни аномалии, будут иметь нормальную продолжительность жизни.
- Низкий рост можно исправить применением гормона роста, но при условии, что у ребенка будет отмечаться дефицит этого гормона
- Как правило, у выживших детей, даже после ряда операций останутся некоторые ограничения в функциональности кистей, например, некоторые из них не смогут сжать пальцы в кулак.
- Стоматологические проблемы встречаются часто.
- Проблемы с почками могут включать нефротический синдром и почечную недостаточность.
- Проблемы с печенью включают врожденный недостаток желчных протоков. Это приводит к прогрессирующему фиброзу и к печеночной недостаточности.
- Гематологические проблемы колеблются от миелодиспластических изменений до острого лейкоза.
Источник

SonoAce-R7
Универсальный ультразвуковой сканер высокого класса, ультракомпактный дизайн и инновационные возможности.
Введение
Синдром выделен в отдельную нозологическую единицу R. Ellis и S. van Creveld в 1940 г. Синдром Ellis – van Creveld (хондроэктодермальная дисплазия, мезодермальная дисплазия, мезодермальная карликовость с шестипалостью) (MIM 225500) – редкое аутосомно-рецессивное состояние с переменными фенотипическими признаками, частота его составляет 1 случай на 60 000 живорождений [1-3]. Синдром изучен достаточно хорошо, в литературе описаны десятки больных с такой патологией. Встречается он у представителей самых разных национальностей и расовых групп. Синдром наиболее распространен среди амишей (США), малочисленной религиозно-этнической общины потомков голландцев-протестантов с большой распространенностью близкородственных браков, что объяснимо, учитывая тип наследования данного заболевания [1-5].
Фенотипический спектр синдрома складывается из: 1) хондродисплазии, 2) полидактилии, 3) эктодермальной дисплазии, 4) пороков сердечно-сосудистой системы.
В названии “хондроэктодермальная дисплазия” отмечены главные особенности синдрома – аномалии хрящевого слоя костей между диафизом и эпифизом, а также дисплазия эктодермальной ткани (ногтей и зубов) [6].
Заболевание характеризуется симметричным укорочением конечностей легкой или средней степени (этот порок отмечен у всех больных), постаксиальной полидактилией, гипоплазией грудной клетки с укорочением ребер средней степени тяжести и пороками сердца (в 60% случаев), чаще всего представленными дефектами межпредсердной перегородки (ДМПП) [6, 7], иногда – единым предсердием, реже – дефектами межжелудочковой перегородки и клапанным стенозом аорты. Полидактилия является постоянным признаком заболевания. Дополнительный палец обычно имеет правильно сформированные пястные и фаланговые кости. Полидактилия на руках отмечается фактически в 80% случаев, на ногах – в существенно меньшем проценте случаев (10-25%). Из других пороков опорно-двигательной системы следует назвать косолапость (до 30% случаев) и вальгусное положение коленей.
Признаками эктодермальной дисплазии являются ногтевая дисплазия, гиподонтия, гипертрофия десен и запоздалое прорезывание зубов. У 70% новорожденных ногти кистей и стоп гипоплазированы, дистальные отделы обычно утолщены. В части случаев ногти могут отсутствовать. Рост волос на голове скудный, вплоть до обширных участков алопеции.
При синдроме Ellis – van Creveld детская летальность отмечается в 50% случаев. Смертность определена степенью выраженности гипоплазии грудной клетки, ведущей к вторичной гипоплазии легких, а также типом и выраженностью порока сердца. Выжившие дети обладают нормальным интеллектуальным развитием, но низким ростом. Окончательный рост от 109 до 162 см. Необходимость коррекции ДМПП не влияет на выживаемость или качество жизни, если преодолены дыхательные проблемы, связанные с грудной гипоплазией. Прочие признаки эктодермальной дисплазии легко поддаются терапии, хотя дефекты прорезывания зубов часто требуют проведения ортодонтических процедур [8]. Обычно нарушена функция кистей, в частности способность сжимать пальцы в кулак.
Пренатальная ультразвуковая диагностика синдрома Ellis – van Creveld потенциально возможна, начиная с середины II триместра беременности (табл. 1). Однако L. Dugoff и соавт. [4] сообщили об ультразвуковой диагностике синдрома в 12 нед.
Таблица 1. Сроки пренатального обнаружения синдрома Ellis – van Creveld по данным литературы.
| Авторы | Срок диагностики, нед |
|---|---|
| F. Cuillier [6] | 24 |
| N. Venkat-Raman и соавт. [9] | 18 |
| C. Sergi и соавт. [10] | 25 |
| T. Tongsong, P. Chanprapaph [11] | 27 |
| I. Torrente и соавт. [12] | 22 |
| F. Qureshi и соавт. [13] | 22-23 |
| О.A.R.M. Al-Asali [14] | 24 |
В отечественных журналах, посвященных проблемам пренатальной ультразвуковой диагностики, мы не встретили ни одного сообщения о дородовом выявлении данного синдрома. В связи с этим представляем собственный опыт пренатальной диагностики синдрома Ellis – van Creveld.
Клиническое наблюдение
Беременная 35 лет, данная беременность первая. Мужу 37 лет, брак не родственный, производственных вредностей супруги не имеют. На учете в женской консультации пациентка состояла с 17 нед беременности. Ультразвуковое исследование проводилось неоднократно (в сроки 17, 23, 28, 32 нед). В сроки 17 и 23 нед беременности в проколах ультразвуковых обследований указано на отсутствие патологии у плода, в срок 28 нед отмечено уменьшение длины трубчатых костей, поставлен диагноз гипохондроплазии. На ультразвуковое исследование 2-го уровня женщина не была направлена. В срок 32 нед повторно отмечено укорочение конечностей, повторно поставлен диагноз гипохондроплазии.
Впервые в медико-генетическое отделение МОНИИАГ женщина обратилась при сроке беременности 37 нед. Ультразвуковое обследование проводилось на сканере Medison Accuvix-XQ с использованием режима поверхностной объемной реконструкции 3D/4D.
При эхографии в 37 нед беременности на фоне многоводия был обнаружен один живой плод мужского пола с аномальным внешним видом. Все длинные трубчатые кости были укорочены и соответствовали 26 нед гестации (рис. 1).
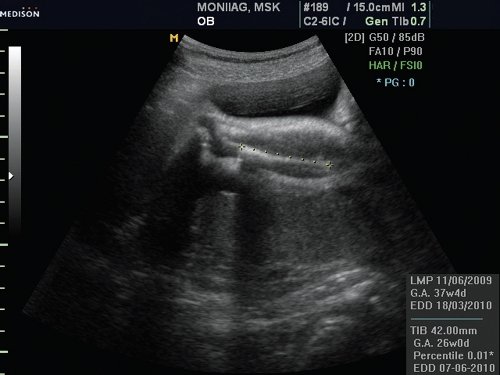
Рис. 1. Укорочение костей голени (микромелия).
Грудная клетка была гипоплазирована. Ее размеры резко диссоциировали с размерами живота плода (рис. 2, 3).
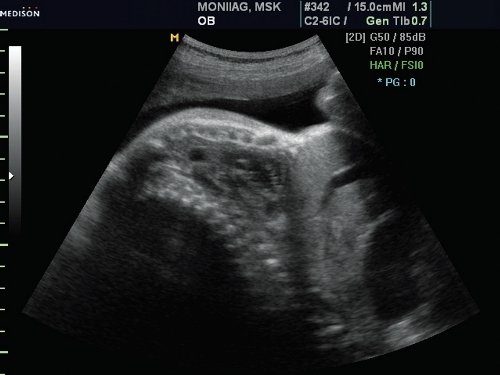
Рис. 2. Гипоплазия грудной клетки.
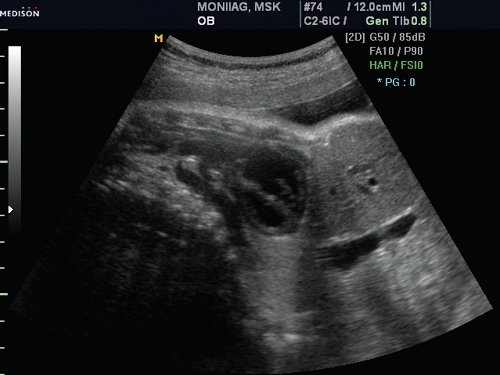
Рис. 3. Гипоплазия грудной клетки.
При осмотре обращало на себя внимание положение головы плода – шейный отдел был в резком разгибании, несмотря на наличие многоводия (рис. 4). Дополнительных полупозвонков не было найдено.
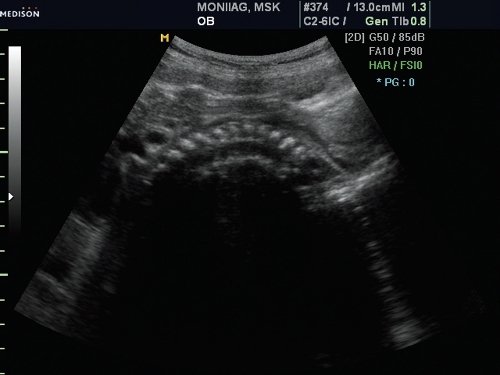
Рис. 4. Гиперразгибание шейного отдела позвоночника.
При обследовании конечностей плода диагностирована постаксиальная полидактилия на одной из стоп плода, что подтверждено в режиме 3D (рис. 5, 6).
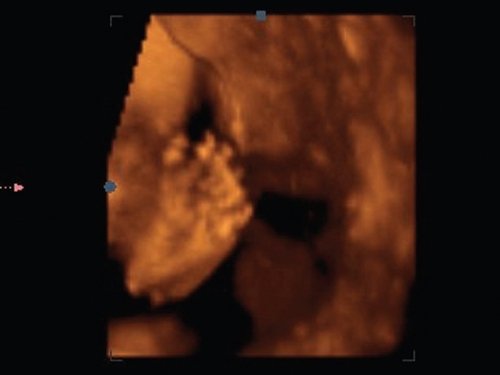
Рис. 5. Полидактилия пальцев стопы, режим 3D.
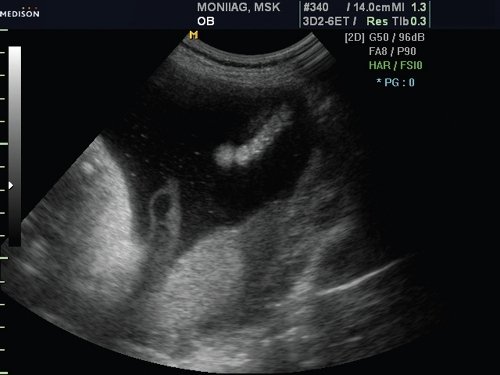
Рис. 6. Полидактилия пальцев стопы, режим 2D.
При осмотре 4-камерного среза сердца плода было выявлено отсутствие межпредсердной перегородки – единое предсердие (рис. 7).
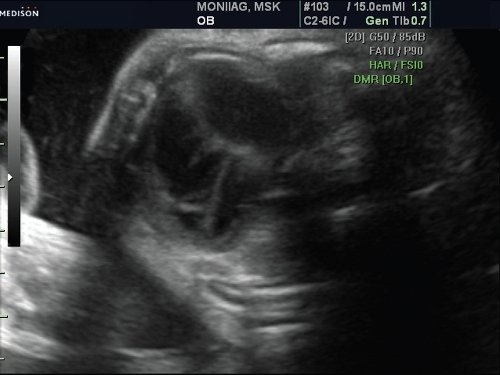
Рис. 7. 4-камерный срез сердца плода. Врожденный порок сердца. Единое предсердие.
Совокупность найденных ультразвуковых признаков патологии позволила нам установить пренатальный диагноз: “37 нед. Множественные врожденные пороки развития плода: скелетная дисплазия (микромелия), гипоплазия грудной клетки, постаксиальная полидактилия. Врожденный порок сердца – единое предсердие. Многоводие”.
Проведено медико-генетическое консультирование с учетом срока беременности. Даны рекомендации о проведении родов в специализированном учреждении. Роды произошли в срок 39 нед. Родился живой мальчик весом 2800 г, длиной 48 см, оценка по шкале Апгар 5/5 баллов. Ребенок консультирован генетиком. Проведены рентгенография грудной клетки (рис. 8), эхография сердца новорожденного. Кариотип новорожденного 46,ХY.
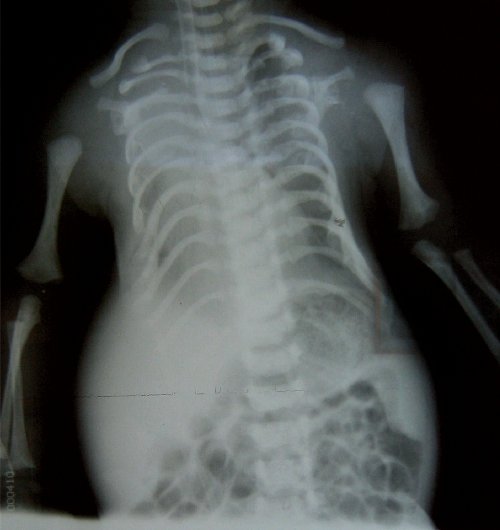
Рис. 8. Рентгенограмма грудной клетки.
При осмотре дополнительно к пренатальным признакам были выявлены облитерация верхней части преддверия рта, гипертрофия десен, готическое небо. Пренатальный диагноз был подтвержден (рис. 9, 10).
Рис. 9. Внешний вид новорожденного с синдромом Ellis – van Creveld.
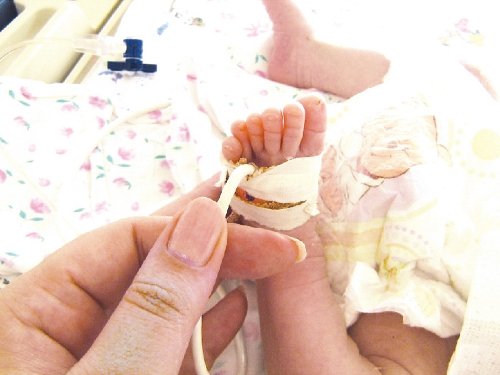
Рис. 10. Полидактилия стопы новорожденного.
С первых часов жизни отмечалась выраженная легочная недостаточность, осложненная наличием врожденного порока сердца. Ребенок был переведен на ИВЛ, умер на 8-е сутки жизни от нараставшей легочно-сердечной недоста точности. При патологоанатомическом исследовании врожденный порок сердца подтвержден.
Обсуждение
Пренатальная ультразвуковая диагностика синдрома Ellis – van Creveld основана на обнаружении симметричного укорочения конечностей, постаксиальной полидактилии, гипоплазии грудной клетки с укорочением ребер и порока сердца. Следует отметить, что в данном случае разными врачами ультразвуковой диагностики не были диагностированы такие ультразвуковые признаки синдрома, как гипоплазия грудной клетки, полидактилия и врожденный порок сердца. В протоколах ультразвуковых обследований доктора отмечали лишь укорочение длины костей. Особое недоумение вызывает то, что ни один из врачей не отметил выраженной гипоплазии грудной клетки. А именно наличие всех перечисленных выше признаков позволяет проводить синдромальный анализ симптомов с формулировкой грамотного пренатального генетического диагноза, который и определяет прогноз не только для жизни, но и для здоровья будущего ребенка.
Однако перечисленные признаки, как в изолированном, так и в сочетанном варианте, встречаются при целом ряде синдромов. Так, при их обнаружении необходимо проводить дифференциальную диагностику с другими состояниями, сопровождающимися “короткими ребрами”. К этим состояниям относятся синдромы коротких ребер – полидактилии – Majewski (type II) и торакоасфиксической дистрофии (синдром Jeune) [15]. Степень выраженности гипоплазии грудной клетки и микромелии при данных состояниях будет различной. Полидактилия может встречаться при всех перечисленных выше синдромах. Самым отличающим эти синдромы признаком является наличие “специфических” сочетанных пороков. При синдроме Ellis – van Creveld – это врожденный порок сердца (пороки межпредсердной перегородки), при синдроме Jeune – это порок развития почек (кистозная дисплазия), при синдроме Majewski – это пороки нижних конечностей, чаще всего проявляющиеся аплазией либо резким укорочением большой берцовой кости и выраженные лицевые дизморфии (табл. 2).
Следует также отметить, что три перечисленных синдрома имеют аутосомнорецессивный путь наследования. Так что, учитывая объективные сложности, которые могут возникнуть при дифференциальной диагностике этих синдромов, при консультировании семьи грамотный доктор не допустит ошибки в заключении и предупредит семью о 25% риске повтора данного заболевания при следующей беременности и, естественно, о возможностях пренатальной ультразвуковой диагностики.
Таблица 2. Дифференциальная диагностика синдрома Ellis – van Creveld.
| Дифференциальные признаки | Синдром Ellis – van Creveld | Синдром Majewski | Синдром Jeune |
|---|---|---|---|
| Укорочение конечностей | ++ | +++ | + |
| Гипоплазия грудной клетки | + | +++ | +++ |
| Полидактилия | Постаксиальная +++ | Пре- и постаксиальная синдактилия +++ | Постаксиальная + |
| Врожденный порок сердца | ++ | – | – |
| Кистозная дисплазия почек | – | ++ | +++ |
| Эктодермальная дисплазия | +++ | – | – |
| Аплазия (резкая гипоплазия) большой берцовой кости | – | +++ | – |
Литература
- Козлова С.И., Семанова Е., Демикова Н.С., Блинникова О.Е. Наследственные синдромы и медико-генетическое консультирование: Справочник. 2-е изд. дополн. М.: Практика, 1996.
- Baujat G., Le Merrer M. Ellis – van Creveld syndrome // Orphanet J. Rare Dis. 2007. V. 4. N2. Р. 27.
- Jenkins S., Morrell D.S. Ellis – van Creveld syndrome: case report and review of the literature // Cutis. 2009. V. 83. N6. Р. 303-305.
- Dugoff L., Thieme G., Hobbins J.C. First trimester prenatal diagnosis of chondroectodermal dysplasia (Ellis – van Creveld syndrome) with ultrasound // Ultrasound Obstet. Gynecol. 2001. V. 17. N1. P. 86-88.
- Ghanekar J., Sangrampurkar S., Hulinaykar R. et al. Ellis – van Creveld syndrome // J. Assoc. Physicians. India. 2009. V. 57. N7. P. 532-534.
- Cuillier F. Ellis – van Creveld syndrome. 2004-11-09-12 // www.thefetus.net
- Белозеров Ю.М. Детская кардиология (наследственные синдромы). Элиста: ЗАОр “НПП “Джангар”, 2008.
- Волков А.Е, Дженти Ф., Медведев М.В. Врожденные пороки опорно-двигательной системы // Пренатальная эхография. Дифференциальный диагноз и прогноз. М.В. Медведев. 2-е изд., перераб. М.: Реал Тайм, 2009. С. 358-359.
- Venkat-Raman N., Sebire N.J., Murphy K.W. et al. Increased first-trimester fetal nuchal translucency thickness in association with chondroectodermal dysplasia (Ellis – van Creveld syndrome) // Ultrasound Obstet. Gynecol. 2005. V. 25. N4. P. 412-414.
- Sergi C., Voigtlander T., Zoubaa S. et al. Ellis – van Creveld syndrome: a generalized dysplasia of enchondral ossification // Pediatr. Radiol. 2001. V. 31. N4. Р. 289-293.
- Tongsong T., Chanprapaph P. Prenatal sonographic diagnosis of Ellis – van Creveld syndrome // J. Clin. Ultrasound. 2000. V. 28. N1. Р. 38-41.
- Torrente I., Mangino M., De Luca A. et al. Firsttrimester prenatal diagnosis of Ellis – van Creveld syndrome using linked microsatellite markers // Prenat. Diagn. 1998. V. 18. N5. Р. 504-506.
- Qureshi F., Jacques S.M., Evans M.I. et al. Skeletal histopathology in fetuses with chondroectodermal dysplasia (Ellis – van Creveld syndrome) // Am.J. Med. Genet. 1993. V. 45. N4. Р. 471-476.
- Al-Asali O.A.R.M. Ellis – van Creveld syndrome. 2008-08-18-09 // www.thefetus.net
- Chen H. Ellis – van Creveld Syndrome: Differential Diagnoses & Workup // www.emedicine.medscape.com/article/943684-diagnosis
SonoAce-R7
Универсальный ультразвуковой сканер высокого класса, ультракомпактный дизайн и инновационные возможности.
Источник