
Синдром ларсона что это такое

Синдром Ларсена – редкое генетическое нарушение, которое связано с развитием широкого спектра различных аномалий и проявлений. Характерные признаки расстройства включают вывихи крупных суставов, пороки развития скелета, отличительные черты лица и проблемы в функциональности конечностей. Дополнительные проявления могут включать в себя ненормальное искривление позвоночника, косолапость, низкорослость и дыхательные трудности. Классическая форма синдрома Ларсена вызывается мутациями в гене FLNB. Мутация может произойти спонтанно или она может быть унаследована по аутосомно-доминантному признаку.
FLNB-связанные расстройства являются группой заболеваний (в том числе аутосомно-доминантный синдром Ларсена), которые развиваются из-за мутаций в гене филамина B (FLNB). Расстройства этой группы характеризуются скелетными аномалиями, которые развиваются в костях рук и ног, в костях позвоночника (позвонки), дислокациями и отличительными чертами лица. Конкретные симптомы и проявления, а также тяжесть этих нарушений могут сильно различаться даже среди членов одной семьи.
Синдром Ларсена. Причины
Классической форме синдрома Ларсена харрактерно аутосомно-доминантное наследование. Доминирующие генетические расстройства возникают тогда, когда только одной копии аномального гена будет необходимо для развития появлений болезни. Ненормальный ген может быть унаследован от обоих родителей, или он может быть результатом новой мутации. Риск передачи аномального гена от родителей к потомству составляет 50 процентов для каждой беременности, независимо от пола будущего ребенка.
Исследователи уже установили то, что классический синдром Ларсена развивается из-за мутаций в гене филамина B (FLNB), который расположен на коротком плече хромосомы 3 (3p14). Каждая хромосома имеет короткое плечо «р» и длинное плечо «q». Дополнительно, исследователи могут указывать конкретные места в хромосоме, которые визуально можно разделить на темные и светлые полосы вдоль каждого плеча. Например, запись сектора 3p14, мутации в котором приводят к развитию этого синдрома, расшифровуется как полоса 14 на коротком плече хромосомы 3. Эти пронумерованные полосы могут точно указать на местоположение генов, которые расположены в этом районе хромосомы.
Ген FLNB содержит инструкции для создания (кодирования) белка, известного как филамин B, который играет роль в правильном развитии внутреннего цитоскелета клеток. Мутации в гене FLNB приводят к тому, что этот ген будет производить белок, но он будет дисфункциональным. Точные функции филамина В и как его дисфункции еще не полностью изучены, но он точно играет важную роль в развитии костной системы и ее соединений.
Некоторые исследователи предполагают, что у некоторых лиц с этим синдромом может наблюдаться мозаицизм. В данном случае, тяжесть заболевания будет зависить от процентного содержания клеток с дефектным геном, и этот сценарий является менее серьезным, чем тот, когда у других людей имеются мутации во всех клетках.
Синдром Ларсена. Эпидемиология
Синдром Ларсен развивается как у мужчин так и у женщин в равном соотношении. По оценкам, этот синдром развивается у 1 из 100000 человек. Из-за трудностей в диагностике синдрома Ларсена, определить истинную частоту будет трудно. Синдром Ларсена был впервые описан в медицинской литературе в качестве самостоятельного расстройства доктором Лореном Ларсеном в 1950 году.
Синдром Ларсена. Симптомы и проявления
Симптомы и тяжесть синдрома Ларсена сильно различаются, в том числе между лицами, принадлежащими к одной и той же семье. В одной большой семье, члены которые имели синдром Ларсена, имели аномалии нёба и несколько дислокаций крупных суставов, в то время как у других членов семьи, с этим синдромом, вообще не было никаких серьезных аномалий, кроме низкорослости и мягких черт, таких как короткие дистальные фаланги и дополнительные кости в запястье и в лодыжке. Что касается низкорослости, то она является самым распространенным проявлением, она встречается в 70% случаев.
Аномалии суставов с характерными чертами лица являются вторыми наиболее частыми проявлениями, связанными с классическим, аутосомно-доминантным синдромом Ларсена. Некоторые симптомы, связанные с синдромом Ларсена присутствуют при рождении, такие как дислокация крупных суставов (80% – бедра, 80% – колени и 65% – локти) и подвывих плеча. Косолапость присутствует у 75% пациентов. Кроме того, суставы пациентов с синдромом Ларсена могут быть чрезвычайно слабым, что делает их более склонными к дислокации. Пальцы могут быть короткими и широкими, с квадратными или закругленными кончиками. Дополнительные кости могут присутствовать в запястьях и лодыжках, а некоторые из этих костей могут сливаться вместе, в детстве.
Аномалии позвоночника встречаются у 84% лиц с синдромом Ларсена, в том числе аномальная боковая кривизна позвоночника (сколиоз) или передняя/задняя кривизна позвоночника в области шеи (шейный кифоз). Лица с синдромом Ларсена и с дисплазиями позвоночника имеют значительный риск развития повреждения мозга и паралича, вторично к повреждению, который происходит, по крайней мере у 15% больных.
Люди с синдромом Ларсена также имеют отличительные черты лица, глаз. Эти черты включают гипертелоризм, широкий лоб и депрессию переносицы. Средняя часть лица может выглядеть плоской. Волчья пасть или расщелины в мягких тканях также могут возникать у 15% лиц. Глухота часто, как правило, предшествует звону в ушах, а проводящий глухота может быть связана с пороками развития косточек среднего уха (у 21% лиц).
Некоторые лица, с классическим синдромом Ларсена, развивают аномальное смягчение хряща трахеи, это состояние известно как трахеомаляция.
Многие люди, которые были описаны в медицинской литературе, имели более тяжелые формы синдрома Ларсена. Эти люди также развивали проблемы с обучаемостью, задержки развития, угрожающие жизни респираторные аномалии и пороки сердца.
Синдром Ларсена. Диагностика
Постановка диагноза синдрома Ларсена проводится на основе тщательного клинического обследования, изучении подробной истории пациента и на основе идентификации характерных клинических и рентгенологических данных. Радиографической экспертизой можно выявить наличие и тяжесть сопутствующих скелетных аномалий. А молекулярно-генетическим тестированием можно окончательно подтвердить наличие мутаций в гене FLNB.
Пренатальная диагностика синдрома Ларсена возможна при применении УЗИ. Более того, скелетные пороки, такие как гиперэкстензии и раздвоенные плечевые кости, определенные черты лица, включая депрессии спинки носа, широко разделенные глаза, выпуклый лоб и аномалии в пальцах могут быть теми признаками, которые смогут навести на синдром Ларсена, хотя и другие генетические расстройства также могут проявлять эти знаки. При достаточно высоком подозрении, может быть выполнено секвенирование гена FLNB, чтобы можно было определить мутацию и прийти к окончательной постановке диагноза. Чтобы не прерывать беременность ребенком с синдромом Ларсена, врачи могут отрекомендовать кесарево сечение для предотвращения травм в конечностях и в шейном отдела позвоночника, во время вагинальных родов.
Синдром Ларсена. Лечение
Лечение синдрома Ларсена направлено на конкретные симптомы и аномалии, которые могут проявляться в каждом человеке по разному. Лечение может потребовать скоординированных усилий команды специалистов. Педиатры, хирурги-ортопеды, черепно-лицевые специалисты и генетики, могут оценить и приступить к лечению ребенка.
Ортопедическая хирургия может быть рекомендована для коррекции скелетных дислокаций или уродств. Физическая терапия может быть необходимой для усиления пораженных суставов. Лечение аномалий суставов часто длительное. Стабилизация шейного отдела позвоночника может быть необходимой в некоторых случаях и она может включать в себя операцию на позвоночнике.
Из-за деформации шейного отдела позвоночника, особое внимание заслуживает интубация (размещение дыхательной трубки в ротовой полости или в носовых полостях во время анестезии), которая может быть необходимой при проведении многочисленных операций.
Для лечения скелетных пороков развития, физическая и профессиональная терапия могут быть необходимы как до так и после операции. Восстановительная хирургия подходит для исправления формы носа и нёба. Дыхательные (респираторные) проблемы могут потребовать применения поддерживающих терапий.
Другие лечебные методики только симптоматические и поддерживающие.
Источник
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РЕСПУБЛИКИ БЕЛАРУСЬ
БЕЛОРУССКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ
КАФЕДРА ТРАВМАТОЛОГИИ И ОРТОПЕДИИ
Е.Р. Михнович
ОСТЕОХОНДРОПАТИИ
Методические рекомендации
ОБЩАЯ ХАРАКТЕРИСТИКА ОСТЕОХОНДРОПАТИЙ
Остеохондропатии (osteochondropathii; от греч. osteon – кость, chondros – хрящ, pathos – страдание; синонимы: остеохондрит, эпифизионекроз, остходролиз, асептический некроз костей) – это особая группа заболеваний костно-суставного аппарата с характерными клинико-рентгенологическими симптомами, в основе которых лежит асептический некроз губчатой костной ткани в местах повышенной механической нагрузки.
Как самостоятельное заболевание остеохондропатия впервые выделена в 1923 г. Аксхаузеном, затем эту патологию описал в 1927 г. Бергман. Однако еще раньше, в 1909 г., асептический некроз головки бедренной кости был описан Леггом, а в 1910 г. – Пертесом.
Классификация остеохондропатий
В зависимости от локализации патологического процесса выделяют 4 группы остеохондропатий:
I. Остеохондропатии эпифизарных концов трубчатых костей: 1) головки бедренной кости (болезнь Легга-Кальве-Пертеса); 2) головки II, реже III, плюсневой кости (болезнь Фрейберга-Келера II); 3) грудинного конца ключицы (болезнь Фридриха); 4) акромиального конца ключицы (болезнь Кливза); 5) головки плечевой кости (болезнь Хасса); 6) головки лучевой кости (болезнь Нильсона); 7) локтевого отростка (болезнь О’Коннора); 8) дистального эпифиза локтевой кости (болезнь Бернса); 9) шиловидного отростка локтевой кости (болезнь Мюллера); 10) головок пястных костей (болезнь Дитерикса); 11) проксимальных эпифизов фаланг пальцев кисти (болезнь Тиманна).
II. Остеохондропатии коротких губчатых и сесамовидных костей: 1) тела позвонка (болезнь Кальве, или плоский позвонок); 2) надколенника (болезнь Синдинга-Ларсена); 3) ладьевидной кости стопы (болезнь Келера I); 4) сесамовидной кости I плюснефалангового сустава (болезнь Ренандера-Мюллера); 5) полулунной кости (болезнь Кинбека); 6) ладьевидной кости кисти (болезнь Прайзера); 7) большой многоугольной кости (болезнь Хармса); 8) крючковидной кости (болезнь Фогеля); 9) гороховидной кости (болезнь Шмира).
III. Остеохондропатии апофизов (апофизиты): 1) апофизов тел позвонков (болезнь Шойерманна-Мау, или юношеский кифоз); 2) гребней подвздошных костей (болезнь Муше-Сорреля-Стефани); 3) лонно-седалищного сочленения (болезнь Ван Нека); 4) бугристости большеберцовой кости (болезнь Осгуда-Шлаттера); 5) бугра пяточной кости (болезнь Хаглунда-Шинца); 6) бугристости V плюсневой кости (болезнь Изелина).
- Частичные клиновидные остеохондропатии суставных поверхностей (osteochondritis dissecans, рассекающий остеохондроз): 1) головки и дистального эпифиза бедренной кости (болезнь Кенига); 2) головки плеча; 3) дистального эпифиза плечевой кости (болезнь Паннера); 4) тела таранной кости (болезнь Диаза).







2.
















Классификация
Поскольку в литературе часто встречается использование дублирующих терминов, неточности или разночтения при переводе фамилий авторов, а также отнесение к ОХП заболеваний, близких к ним по клинической картине, но имеющих иной генез, классификации следует уделить особое внимание.
По анатомической локализации, течению и прогнозу ОХП подразделяются на четыре группы.
1. Остеохондропатии эпифизарных отделов трубчатых костей (спонтанный, или идиопатический, асептический некроз):
- проксимального эпифиза (головки) плечевой кости – болезнь Хасса (Гасса);
- дистального эпифиза локтевой кости – синдром Бернса;
- шиловидного отростка локтевой кости – болезнь Мюллера;
- головки лучевой кости – болезнь Нильсона;
- гороховидной кости – болезнь Шнира;
- пястных костей и оснований фаланг пальцев рук и ног – синдром Тиманна (Тимана), или болезнь Тиманна-Флейшнера;
- головок пястных костей – болезнь Дитриха;
- головки бедренной кости, или остеохондрит головки бедра, или псевдококсалгия – болезнь Пертеса (Легга-Кальве-Пертеса);
- медиального мыщелка большеберцовой кости, или tibia vara, или нерахитическая саблевидная голень у детей – синдром Блаунта-Барбера (болезнь Блаунта, синдром Блаунта-Эрлахера-Биезиня-Барбера);
- головки II, реже III, IV плюсневой кости – болезнь Келера II, болезнь Келера-Фрейберга (Фрайберга);
- V плюсневой кости – болезнь Излена;
- головки II плюсневой кости – болезнь Фрейберга;
- внутреннего или грудинного конца ключицы – болезнь Фридриха;
- наружного конца ключицы, соединяющегося с лопаткой, – болезнь Кливза;
- крючковидного отростка или кости – болезнь Фогеля.
2. Остеохондропатии губчатых костей:
- тела одного позвонка в нижнегрудном или верхнепоясничном отделе – болезнь Кальве;
- полулунной кости запястья – болезнь Кинбека;
- ладьевидной кости запястья – болезнь Прайзера;
- метакарпальных костей – синдром Дитриха;
- многогранной кости – болезнь Хармса;
- ладьевидной кости стопы – болезнь Келера I;
- эпифиза медиальной клиновидной кости стопы – синдром Бринсона;
- эпифиза промежуточной клиновидной кости стопы – синдром Кюнчера;
- остеохондропатия коленной чашечки – болезнь Ларсена;
- хондропатия, или хондромаляция коленной чашечки – болезнь Левена (синдром Бюдингера-Лудлоффа-Левена, болезнь Хаглунда-Левена-Фрюнда);
- коленной чашечки в сочетании с гидроартрозом – болезнь (синдром) Синдинга-Ларсена-Иогансона;
- верхнего полюса коленной чашечки – болезнь Земмельрока;
- сесамовидной кости I плюснефалангового сустава стопы – болезнь (синдром) Ренандера-Мюллера.
3. Остеохондропатии апофизов:
- юношеский апофизит грудных позвонков, преимущественно VII–X, как хондроили спондилодисплазия, или круглая спина – болезнь Шейерманна (Шойермана) – Мау (юношеский остеохондропатический кифоз, болезнь Шморля);
- спонтанный асептический некроз вертлужной впадины тазовой кости – синдром Хесслера;
- подхрящевой асептический некроз лобковой кости – болезнь Пирсона;
- остеохондропатия седалищно-лобкового сочленения – болезнь Ван-Некка, или синдром Одельберга;
- бугристости большеберцовой кости – болезнь Осгуда-Шлаттера;
- эпифиза пяточной кости – болезнь Севера;
- бугра (апофиза) пяточной кости – болезнь Хаглунда (Гаглунда-Шинца).
4. Частичные клиновидные некрозы суставных участков костей (так называемый рассекающий, отсекающий суставной край, или расслаивающий остеохондроз):
- дистального эпифиза плечевой кости – болезнь (синдром) Паннера;
- медиального мыщелка бедренной кости – болезнь Кенига;
- частичный асептический некроз, или травматический спондилит одного грудного позвонка – болезнь (синдром) Кюммеля (Кюммеля-Вернея);
- тела таранной кости – болезнь Хаглунда-Севера, болезнь Диаза-Вейса-Мюллера.
Источник
Наш генетический код настолько сложный, что практически любая серьезная поломка способна вызвать цепную реакцию и отразиться на человеке не с самой лучшей стороны. Ученые постоянно находят новые заболевания, но, по их собственным словам, на девяносто процентов геном остается неисследованным.
Описание
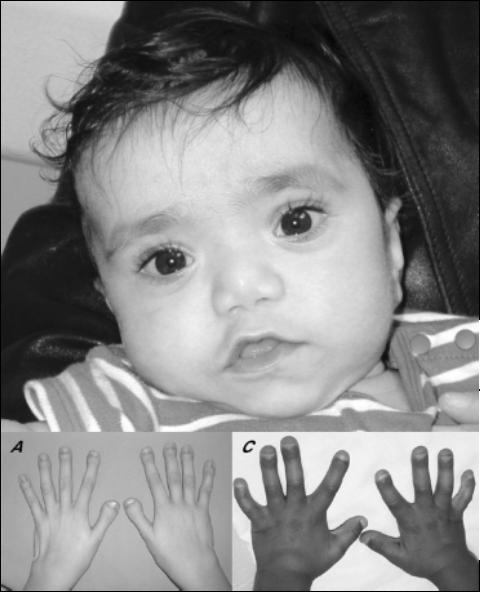
Синдром Ларсена (МКБ 10 – код М89) – это редко встречающееся генетическое заболевание, которое имеет широкий круг фенотипических проявлений. Наиболее характерными признаками считаются вывихи и подвывихи крупных суставов, наличие пороков развития костей лицевого черепа и проблемы с функциями конечностей. К второстепенным проявлениям относят сколиоз, косолапость, низкий рост и трудности с дыханием.
Синдром Ларсена вызывается точечными мутациями, которые могут произойти как спонтанно, так и быть переданными по наследству по аутосомно-доминантному типу. С изменениями в гене FLNB связывают целую группу заболеваний, проявляющихся нарушениями в скелетной системе. Конкретные проявления могут быть разными даже среди родственников.
Причины
Что же должно произойти в формирующемся организме, чтобы возник синдром Ларсена? Причины этого заболевания все еще скрываются в научном полумраке. Известно только, что для него характерным является аутосомно-доминантное наследование. То есть всего одной копии измененного гена будет достаточно, чтобы передать патологию своим детям, а может быть, даже внукам. Ген может быть получен от родителей (от обоих или от одного) либо являться результатом спонтанной мутации. Риск унаследовать данное заболевание – 50/50, независимо от пола ребенка и количества беременностей.
Измененный ген расположен в коротком плече третьей хромосомы. При желании исследователи могут точно указать место, где наследственная информация подверглась изменениям. В норме этот ген кодирует белок, известный в научных кругах как филамин В. Он играет значительную роль в развитии цитоскелета. Мутации приводят к тому, что белок перестает выполнять возложенные на него функции, и от этого страдают клетки организма.
У людей с данным синдромом возможен мозаицизм. То есть тяжесть и количество проявлений заболевания напрямую зависят от того, сколько клеток оказалось поражено. Некоторые люди могут даже не подозревать о том, что имеют дефект этого гена.
Эпидемиология

Синдром Ларсена с одинаковой частотой развивается как у мужчин, так и у женщин. По весьма приблизительным оценкам, данное заболевание встречается у одного новорожденного из ста тысяч. Это, по счастью, большая редкость. Оценки считаются недостоверными, потому что существуют определенные трудности в выявлении этого синдрома.
Впервые в медицинской литературе заболевание было описано в середине двадцатого века. Лорен Ларсен и соавторы нашли и зафиксировали шесть случаев проявления синдрома у детей.
Симптомы
Синдром Ларсена, как уже упоминалось выше, может по-разному проявляться даже между близкими родственниками. Наиболее характерными признаками заболевания считаются изменения костей лица. К ним относятся: широкая низкая переносица и широкий лоб, плоское лицо, наличие незаращения верхней губы или твердого неба. Кроме того, у детей имеются дислокации крупных суставов (бедренного, коленного, локтевого) и подвывихи плеча.
Пальцы у таких людей короткие, широкие, со слабыми разболтанными суставами. В запястьях могут присутствовать дополнительные кости, которые с возрастом сливаются и нарушают биомеханику движений. У некоторых людей наблюдается такой редкий феномен, как трахеомаляция (или размягчение хрящей трахеи).
Диагностика
Диагноз “синдром Ларсена” ставится только после полного обследования больного, тщательного изучения его истории болезни и наличия характерных рентгенологических симптомов. Кроме того, полноценная радиографическая экспертиза может выявить и сопутствующие аномалии развития скелета, которые имеют косвенное отношение к заболеванию.
Ультразвуковая диагностика еще во внутриутробном периоде может выявить синдром Ларсена. Фото костных образований для хорошо подготовленного специалиста УЗИ может стать отправной точкой для поиска генетических аномалий плода. Так как с первого взгляда нельзя сказать, какая именно болезнь привела к патологии лицевого черепа и костей конечностей, будущей матери рекомендуют сделать амниоцентез и провести генетическую экспертизу для поиска мутации в третьей хромосоме.
Если заболевание подтверждается, но супруги приняли решение о сохранении беременности, то будущей матери рекомендуется провести процедуру кесарева сечения, чтобы не повредить кости ребенка в процессе прохождения его через таз женщины при естественных родах.
Лечение

Лечебные мероприятия направлены не на устранение заболевания, а на снижение клинических проявлений. Этим занимаются педиатры, ортопеды, специалисты по челюстно-лицевой хирургии и генетики. После оценки первоначального состояния ребенка и оценки всех рисков они могут приступить к коррекции имеющихся нарушений.
Самый щадящий вариант, которым лечится синдром Ларсена, – массаж. Он необходим для укрепления мышц и связок, удерживающих суставы, а также для улучшения поддержания спины и выпрямления позвоночника. Но до того как приступить к терапевтическим методам, потребуется ряд операций. Они необходимы для коррекции грубых деформаций скелета или уродств, стабилизации позвонков. При трахеомаляции требуется интубация, а затем и постановка дыхательной трубки (на постоянной основе), которая будет поддерживать проходимость верхних дыхательных путей.
Лечение этого заболевания – процесс длительный, который может растянуться на годы. С взрослением ребенка нагрузка на кости увеличивается, и ему снова может потребоваться физическая реабилитация, лечебная физкультура, а может быть, даже и хирургическое вмешательство.
Источник