
Синдром ломкой х хромосомы фото


Синдром Мартина-Белл – это наследственная болезнь, которая характеризуется стойким интеллектуальным снижением, расстройствами аутистического спектра и специфическими фенотипическими особенностями. Ключевой симптом – недостаточность познавательных функций. Отмечается гиперактивность, дефицит коммуникативных способностей, замкнутость. Лицо удлиненное, ушные раковины большие, лоб выступающий, кончик носа загнутый. Диагностика основывается на клинико-анамнестических данных и результатах биогенетического анализа. Лечение симптоматическое, включает использование медикаментов и психолого-педагогическую коррекцию.
Общие сведения
Синдром Мартина-Белл получил свое название по фамилиям исследователей, впервые описавших патологию. В 1943 году физиологи из Великобритании Д. Мартин и Д. Белл изучали 11 случаев олигофрении у мужчин из одной семьи, в которой женщины имели нормальное интеллектуальное развитие. Генетическая основа заболевания была выявлена в 1969 году американским генетиком Г. Лабсом. Синонимичное название – синдром ломкой X-хромосомы. Распространенность среди мальчиков составляет 1:4 000, среди девочек – 1:6 000. Согласно данным зарубежных врачей-генетиков, частота синдрома Мартина-Белл у пациентов мужского пола с умственной отсталостью достигает 1,9-5,9%. Отечественные исследования указывают на более высокие значения, в соответствии с ними этот синдром имеют 8-10% больных олигофренией.

Синдром Мартина-Белл
Причины
Синдром Мартина-Белл является результатом дефекта гена FMR1, расположенного в X-хромосоме. Наследование происходит по доминантному сцепленному с полом типу с неполной пенетрантностью. У мужчин присутствует одна X-хромосома, поэтому мутантный аллель всегда провоцирует болезнь. У женщин есть две половые хромосомы типа X: одна активная, другая – резервная, инактивированная. Таким образом, при наличии мутации в одном из двух генов FMR1 заболевание проявляется или нет в зависимости от активности измененной хромосомы. Мужчины с ломкой хромосомой X не могут передать ее сыновьям, но передают всем дочерям, которые либо болеют, либо остаются здоровыми носителями мутации. Женщины с дефектной хромосомой передают ее детям обоих полов с вероятностью 50%. Наследование синдрома учащается от поколения к поколению, этот феномен называется парадоксом Шермана.
Патогенез
При секвенировании FMR1-гена было выявлено, что основой симптоматики и цитогенетически определяемой ломкости хромосомы X является многократное увеличение количества единичных тринуклеотидов ЦГГ. Это приводит к подавлению транскрипции и последующему недостаточному производству белка FMR1, ответственного за развитие центральной нервной системы, а именно – за формирование аксонов и синапсов, появление и усложнение нейронных связей, успешность процессов обучения и запоминания.
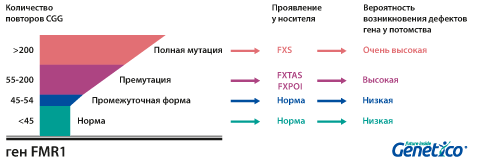
Участок хромосом, подверженный структурным изменениям при наследственном синдроме Мартина-Белл, может находиться в четырех состояниях, характеризующихся различным удлинением повторяющихся последовательностей тринуклеотидов. При отсутствии болезни и носительства определяется нормальное количество повторов – от 6 до 39. В промежуточном состоянии диагностируется 40-60 повторов, в состоянии премутации – 55-200. В обоих случаях заболевание отсутствует. Поскольку экспансия тринуклеотидов возможна лишь в период гаметогенеза, премутация способна превратиться в полную мутацию. Это происходит при передаче измененного материнского гена, аллель «утяжеляется» во время овогенеза. При полной мутации выявляется больше 200 повторов ЦГГ, чаще всего – от 230 до 4 000.
Симптомы
Дети рождаются с увеличенной массой тела, в среднем – 3,5-4 кг. Первыми обращают на себя внимание фенотипические особенности младенцев. Характерен макроорхизм – увеличение яичек без эндокринного заболевания. Окружность головы больше нормы или соответствует ее верхним границам. Лоб высокий и широкий, лицо вытянутое с уплощенной средней частью. Нос имеет слегка клювовидный загиб, ушные раковины крупные, располагаются низко. Суставы отличаются хорошей подвижностью, кости кистей и стоп широкие. Кожа зачастую гиперэластичная, волосы и радужные оболочки глаз светлого оттенка. Фенотипические признаки могут быть выражены по-разному, от одного-двух едва определяемых до полного комплекса.
Ключевое клиническое проявление заболевания – умственная отсталость. Стойкое интеллектуальное снижение проявляется слабым развитием сложных форм мышления и памяти. Пациентам недоступно понимание абстрактно-логических высказываний и явлений, использование категорий, установление аналогий. Сравнение, анализ и обобщение могут осуществляться на простом уровне, например, в конкретных бытовых ситуациях. Словарный запас обеднен. У многих мальчиков IQ равен 40-50 баллам, реже достигает 70-79. Относительно сохранна номинативная речь и зрительное восприятие. У девочек когнитивное снижение менее выраженное, соответствует легкой степени олигофрении или пограничному уровню интеллектуального развития.
Другой типичный симптом заболевания – своеобразие речи. Она ускоренная, сбивчивая, изобилует повторами, эхолалиями и персеверациями. Аутистические расстройства представлены трудностями коммуникации и поведенческими нарушениями. Дети часто проявляют агрессивность и замкнутость при попытке установления контакта. В тяжелых случаях развивается мутизм – полное отсутствие речи как средства общения. В поведении преобладает двигательная расторможенность, гиперактивность, стереотипии, самопровреждения. Пациенты избегают смотреть в глаза, не допускают прикосновений, но по сравнению с больными аутизмом интерес к общению присутствует. Стереотипные движения включают хлопки руками, прыжки, вращения вокруг своей оси, встряхивания руками, бег по кругу, гримасничанье и однообразное хныканье. Имеются трудности планирования и контроля поведения, переключения внимания и пространственной координации.
Неврологические симптомы неспецифичны. Определяется легкое снижение мышечного тонуса, двигательная дискоординация. Недостаточное развитие мелкой моторики затрудняет освоение письма, некоторых игровых и бытовых навыков (сборки конструктора, рисования, шитья и др.). У части больных имеются глазодвигательные нарушения, усиление сухожильных рефлексов, экстрапирамидные паракинезы, например, зажмуривание глаз, нахмуривание бровей, гримасничанье. При тяжелых формах синдрома возникают эпилептические припадки. У 25% пациенток с премутационным состоянием развивается первичная недостаточность яичников.
Диагностика
При выраженных фенотипических изменениях заболевание может быть обнаружено с первых месяцев жизни ребенка – неонатологи и врачи-педиатры обращают внимание на увеличенные размеры яичек и характерные особенности лица. В иных случаях подозрение на умственную отсталость возникает в возрасте от полугода до 2-3 лет. В этот период прослеживается отставание умственного развития, поведенческие и речевые нарушения. Дифференциальная диагностика нацелена на исключение РАС, в частности раннего детского аутизма, а также умственной отсталости другого происхождения (не связанной с ломкостью хромосомы Х). Обследование проводится психиатрами, неврологами и врачами-генетиками, включает:
- Клинический опрос, осмотр. В беседе с ребенком на первый план выходит снижение интеллекта, гиперактивность и расторможенность поведения, нарушение коммуникативных навыков. Уровень психического развития не соответствует возрасту, методики исследования интеллекта выявляют олигофрению (IQ – 40-79 баллов). Внешне наблюдаются характерные фенотипические признаки, при неврологическом осмотре выявляется мышечный гипотонус, усиленные сухожильные рефлексы, паракинезы.
- Генеалогический анализ. В отличие от других форм олигофрении при синдроме Мартина-Белл прослеживается наследственная передача болезни. Как правило, у пациента имеются родственники с данным заболеванием, чаще – мужчины (дед, дядя, брат). Иногда признаки легкого интеллектуального снижения обнаруживаются у матери, но диагноз у нее часто не установлен (не подтвержден).
- Биогенетическое исследование. В лабораторных условиях исследуется строение ДНК: определяется количество ЦГГ-повторов и статус метилирования. Применяется ПЦР и цитогенетический метод. Диагноз подтверждается, если количество триплетных повторов составляет более 200. При результате 60-199 возможны легкие фенотипические проявления болезни, риск развития патологии в следующем поколении (если показатель диагностирован у женщины).
Лечение синдрома Мартина-Белл
Методы специфической терапии синдрома в настоящее время отсутствуют. Проводится симптоматическое медикаментозное лечение и психолого-педагогическая коррекция. Усилия врачей и специальных психологов направлены на минимизацию эмоционально-поведенческих отклонений, овладение навыками ходьбы, речи и общения, чтения и письма. Медикаментозная терапия включает прием психостимуляторов, антидепрессантов, ноотропов, противоэпилептических средств и гормональных препаратов (при первичной недостаточности яичников). Обучение пациентов проводится по специальным коррекционно-развивающим программам. Для улучшения социальных навыков используются методы когнитивно-поведенческой терапии, групповые тренинги.
Прогноз и профилактика
Синдром Мартина-Белла не имеет осложнений и не сокращает продолжительность жизни больных, поэтому при своевременной и адекватной медико-психолого-педагогической помощи прогноз достаточно благоприятный: пациенты осваивают навыки общения и самообслуживания, обучаются в специальных школах, иногда овладевают рабочими профессиями. Профилактика основана на медико-генетическом консультировании пар из групп риска и пренатальной диагностике синдрома. Эти меры необходимы женщинам с синдромом преждевременного истощения яичников, семьям, в которых диагностированы премутационные состояния FMR1 или выявлены случаи интеллектуальной недостаточности у мальчиков и мужчин.
Источник
Генетические особенности синдрома ломкой Х-хромосомы
Среди группы наследственных болезней есть два заболевания, относящихся к самым частым причинам интеллектуальной недостаточности. Самая известная и наиболее распространённая патология – синдром Дауна, связанный с наличием лишней 21-ой хромосомы в геноме человека. В этой статье мы расскажем о втором по распространенности наследственном заболевании, которое приводит к умственной отсталости, а также может сопровождаться другими клиническими проявлениями.
Синдром ломкой X-хромосомы или синдром Мартина-Белл является результатом нарушения в гене FMR1 (fragile X mental retardation-1), который расположен на Х-хромосоме и играет важную роль в появлении и развитии нервных связей, обучении и запоминании. Частота этого синдрома среди мальчиков составляет 1:4000.
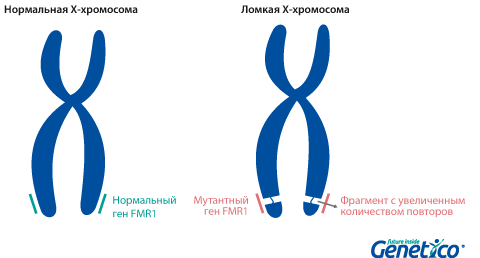
Так называемая «ломкость» X-хромосомы проявляется в том, что хромосома выглядит нетипично при специальном окрашивании, как будто один кусок отделился, хотя физически она остается цельной. Генетическая основа этого явления заключается в увеличении числа тринуклеотидных повторов CGG в гене FMR1, расположенном на X-хромосоме.
У здоровых людей число повторов в этом гене колеблется от 5 до 54. Если повторов больше 200, то наработка белка с гена FMR1 нарушается, что приводит к развитию синдрома Мартина-Белл и клиническому проявлению заболевания. Премутационное состояние — это количество повторов CGG от 55 до 200. В таком состоянии заболевание у людей в типичной форме не проявляется, но чем больше повторов в этом гене у носителя, тем больше вероятность того, что у ее или его детей количество повторов будет больше 200 и заболевание разовьется. В случае носительства премутации при формировании половых клеток количество повторов может увеличиваться, поэтому если у родителя количество повторов от 55 до 200, то высока вероятность рождения ребенка с мутантным геном FMR1 и синдромом Мартина-Белл. При этом носительство премутационного состояния будущим папой и мамой неравнозначно по вероятности возникновения мутантного аллеля у их детей: если носитель – мама, то вероятность значительного увеличения числа повторов гораздо выше. Количество повторов от 45 до 54 является промежуточной формой, которая не имеет никакого влияния на здоровье человека, но может приводить к проблемам у будущих поколений, как и в случае премутационного состояния гена.
Важно учитывать, что наследование и развитие заболевания зависит от пола, так как ген FMR1 находится на Х-хромосоме. У мужчин только одна Х-хромосома, которую они получают от матери. Поэтому, в случае, если эта одна хромосома оказалась «ломкой», у них проявляется заболевание. У женщин две Х-хромосомы, однако активно работает только одна из них. Поэтому наличие одной Х-хромосомы с мутантным геном FMR1 может не проявляться клинически, в случае инактивации именно «ломкой» хромосомы, или приводить к развитию заболевания в 30-50% случаев. Мужчина с ломкой Х-хромосомой может передать её всем дочерям, но ни одному из сыновей. Женщина с мутантной хромосомой имеет шансы передать её как сыновьям, так и дочерям с равной вероятностью.
Премутационное состояние гена влияет как на судьбу потомков носителя такого гена, так и непосредственно на его здоровье:
Развитие первичной недостаточности яичников (FXPOI) (снижение овариального резерва и наступление менопаузы до 40 лет). Мутация FMR1 является причиной преждевременного истощения яичников у 5% женщин с этим диагнозом. Среди носительниц премутации примерно у четверти развивается это состояние. Оно влияет не только на общие репродуктивные возможности, но и на подбор протокола стимуляции при ВРТ, так как часто оказывается причиной бедного ответа яичников на стимуляцию. Интересно, что по данным, полученным в центре Genetico, хотя бедный ответ яичников на стимуляцию влияет на число получаемых в цикле эмбрионов, он не приводит к увеличению доли анеуплоидных эмбрионов.
Тремор/атаксия, ассоциированные с ломкой Х-хромосомой (FXTAS). Это состояние чаще развивается у мужчин: при носительстве премутации мужчиной проявляется в 33% случаев, а при носительстве премутации женщиной – лишь в 5-10%. Синдром FXTAS начинает проявляться в пожилом возрасте. Наблюдается тремор, шаткая походка, может страдать речь.
Метод диагностики, используемый в лаборатории Genetico, основан на использовании полимеразной цепной реакции с особым набором праймеров, позволяющих не только детектировать нормальное, премутационное и мутационное состояния, но и точно определить количество повторов в случаях, когда их меньше 200. Такая диагностика позволяет выявить синдром ломкой X-хромосомы на молекулярном уровне, а также оценить вероятность рождения ребенка с этим синдромом и возможность развития у пациента расстройств, связанных с увеличенным количеством повторов в гене FMR1. Такая диагностика также позволяет детектировать наличие AGG повторов среди повторов CGG. Полагают, что участки AGG, прерывающие длинную последовательность из CGG повторов, придают ДНК устойчивость и снижают риск увеличения количества повторов в следующем поколении.
Генетический тест, определяющий количество повторов в гене FMR1, рекомендуется пройти в первую очередь женщинам с синдромом преждевременного истощения яичников или с выявленной неслучайной инактивацией Х-хромосомы (косвенный признак), семьям, в которых есть сыновья с интеллектуальной недостаточностью. Также анализ состояния гена FMR1 необходим:
1) женщинам с репродуктивными проблемами или нарушениями фертильности, связанными с повышенным уровнем фолликулостимулирующего гормона (ФСГ)
2) пациентам с интеллектуальной недостаточностью и их родственникам
3) тем, у кого в семье были случаи синдрома ломкой Х-хромосомы или умственной отсталости без точного диагноза
4) женщинам, у родственников которых наблюдались нарушения, связанные с премутационным состоянием FMR1
5) пациентам с поздно проявившимся тремором и мозжечковой атаксией (нарушения согласованности работы мышц из-за поражения систем мозга, управляющих движением мышц).
В случае обнаружения бессимптомного носительства мутации в гене FMR1 у женщины может быть рекомендовано использование донорских ооцитов или проведение преимплантационной генетической диагностики (ПГД) с целью исключить возможность проявления синдрома у ребенка. Также важно правильно оценивать риск рождения больного ребенка в случае премутационного состояния гена FMR1 у будущих родителей. В таком случае по результатам теста рекомендуется консультация врача-генетика.
Автор: Очир Мигяев
Стажер лаборатории Genetico
Источник

Синдром Мартина-Белла (СМБ, синдром ломкой X-хромосомы) — передающееся по наследству заболевание, основным клиническим признаком которого является умственная отсталость. Это генетическое нарушение обусловлено ломкостью дистального плеча Х-хромосомы — его резким сужением. Впервые о синдроме заговорили в 1943 году. И лишь спустя 50 лет группой ученых был обнаружен ген, мутация которого приводит к развитию болезни.
Генетики из Ирландии и Англии Д. Мартин и Д. Белл в начале 20 века описали семью, в которой у абсолютно здоровых матерей рождались умственно отсталые сыновья. Причем данный дефект развития нервной системы наследовался сцеплено с полом. Благодаря этим докторам синдром и получил свое название. Спустя несколько лет ученые, проводя цитогенетическое обследование, выявили ген, мутация которого приводит к образованию вторичной перетяжки на длинном плече Х-хромосомы. Обнаружение первого пренатального маркера синдрома дало возможность пациентам принять правильное решение о сохранении или прерывании беременности.

Мальчики страдают данной генетической аномалией в 3 раза чаще, чем девочки. У них заболевание протекает намного тяжелее. Это связано наличием второй Х – хромосомы в кариотипе представительниц слабого пола, компенсирующей патологические изменения. Болезнь эта довольно распространенная: на 4000 новорожденных мальчиков рождается 1 больной. На возникновение недуга не оказывают влияния национальность, цвет кожи и разрез глаз, экологические, материальные и социально-бытовые условия. Каждый пятый мужчина, рожденный с измененным геном, является его носителем и считается клинически здоровым. Все остальные имеют признаки умственной отсталости различной степени выраженности: от легкой до тяжелой.
СМБ — относительно новое заболевание, имеющее высокий процент детской инвалидности. Нарушение психофизического развития неуклонно прогрессирует с самого раннего детства. Синдром имеет код по МКБ-10 Q99.2 и наименование «Ломкая Х-хромосома».
Этиопатогенез
СМБ — генетическая мутация, приводящая к истончению определенного участка Х-хромосомы. Ген, отвечающий за появление такой хрупкости, полностью или частично прекращает продукцию специфического белка, обеспечивающего нормальное функционирование нервной ткани. У здоровых людей этот протеин играет важную роль в процессе обучения и запоминания. Его дефицит заканчивается развитием умственной отсталости.

У лиц с СМБ мутация гена приводит к изменению молекулярной структуры ДНК. Она перестраивается и становится нестабильной. Экспрессия гена фактически прекращается. Наследственная информация в виде последовательности нуклеотидов перестает преобразовываться в функциональный продукт — РНК или белок.
В кариотипе мужчин имеется лишь одна Х-хромосома (46 ХУ). Если она содержит мутантный ген, то у носителя всегда проявится болезнь. Женщины имеют две Х-хромосомы (46 ХХ), поэтому часто остаются здоровыми. Вторая X-хромосома как бы компенсирует имеющийся дефект. Такие женщины обычно эмоционально неустойчивы, страдают депрессиями и фобиями. Мужчины-носители пораженной Х-хромосомы передают ее своим дочерям, а женщины с одинаковой вероятностью девочкам и мальчикам. Половые хромосомы состоят из цепочек аминокислот, которые повторяются с определенной периодичностью и в определенной последовательности. Результатом патологического увеличения таких повторов является истончение участка Х-хромосомы.
В норме количество повторов нуклеотидов колеблется от 29 до 31. У больных с СМБ встречаются разные варианты:
- 40-60 повторов – промежуточное состояние: отсутствие клинических проявлений, передачи синдрома по наследству через несколько поколений.
- 55-200 повторов — премутация с неизмененной структурой гена. Носители пораженной хромосомы не имеют видимых изменений нервной системы. Возможно развитие в старческом возрасте атаксии, тремора, амнезии, деменции и когнитивных расстройств. Вероятность передачи синдрома очень высока. Отцовская передача премутационных аллелей дочерям не проявляется клиническими симптомами патологии. Если имеется материнская передача, развивается синдром с характерными проявлениями.
- Увеличение повторов до 4000 приводит к дисфункции особого гена, отвечающего за правильное психоэмоциональное развитие. Первые клинические проявления у больных детей появляются не сразу, а спустя некоторое время после рождения. Нарушение психомоторного развития неуклонно прогрессирует. У будущих поколений недуг будет протекать все тяжелее.
Диагностикой синдрома занимаются врачи-генетики. Предположить его наличие можно с помощью электроэнцефалографии, поскольку у больных обнаруживается сходная биоэлектрическая активность мозга. Для постановки окончательного диагноза необходимы специальные методы обнаружения генетических аномалий. В настоящее время появилась возможность не только диагностировать эту патологию, но и лечить. Современная фармацевтическая промышленность разработала и выпустила лекарственные препараты, способные улучшить память, внимание и общее состояние больных, а также снизить их двигательную расторможенность.
Симптоматика
Клиника синдрома довольно многообразна, но не все симптомы заболевания проявляются одинаково у разных больных. К основным специалисты относят: снижение интеллектуального уровня, нарушение психоэмоционального развития, физические расстройства.

Клинические признаки синдрома Мартина-Белла возникают не сразу после рождения, а к концу первого года жизни. Первым проявлением патологии является сниженный тонус мышц. Перевозбужденный или пассивный ребенок плохо реагирует на голос окружающих и даже матери. У него развивается гипо- и арефлексия, что проявляется снижением или отсутствием хватательного и сосательного рефлексов. Спустя некоторое время симптомы становятся более очевидными.
- Больные дети поздно начинают ходить и говорить. Они говорят быстро и сбивчиво, слова опережают друг друга. Преобладает бормочущая речь, которая воспринимается с большим трудом. Пациенты часто повторяют целые фразы и предложения, одни и те же слова или их окончания. Некоторые из них заикаются. В тяжелых случаях речевые нарушения достигают мутизма – патологии, при которой человек сохраняет полное молчание.
- Дискоординация движений заключается в хаотичном размахивании руками. Гиперкинетические признаки – подпрыгивания, похлопывания руками, повороты вокруг своей оси, встряхивание кистями, гримасничество, хныканье. Больные не сидят на одном месте, принимают вычурные позы, совершают бессмысленные движения. Задержка психомоторного развития проявляется значительным отставанием ребенка от своих сверстников.
- В характере преобладает упрямство, злость с очень сильными и неконтролируемыми приступами гнева, эмоциональная лабильность, невнимательность и несосредоточенность, необоснованные страхи. Больные дети очень робкие, они боятся шума и избегают скопления людей. Постоянное беспокойство вызвано плачем и истериками. С ними сложно установить зрительный контакт, они не переносят прикосновений.
- Неврологические расстройства — гиперподвижность, нервный тик, частое моргание, тремор, эпиприпадки.
- Ребенка отличает характерный внешний вид — оттопыренные уши низко расположены на большой голове; широкий и высокий лоб занимает большую часть лица; на продолговатом лице расположен крючковидный нос и выступающий вперед подбородок; глаза больных часто косят; широкие ладони и стопы; кожа гиперэластичная и легко растяжимая.
- Эндокринные нарушения имеются у всех больных – лишний вес, крупные яички и яичники с их кистозным перерождением, раннее половое созревание.
- Уровень интеллекта колеблется от небольшой умственной отсталости до ее тяжелых форм. Чаще всего больные являются олигофренами.
- Врожденные патологии внутренних органов — пороки сердца, высокое дугообразное небо, плоскостопие, малоподвижность суставов, деформация стоп и голеней, кривые ноги.
- Сопутствующие заболевания — постоянный отит, приступы шизофрении.
У женщин гипофункция яичников приводит к раннему наступление климактерического периода, исчезновению менструаций и появлению вегетативных признаков. Чтобы остановить дальнейшее прогрессирование патологии, требуется проведение заместительной гормональной терапии.
Поскольку симптомы болезни напоминают аутизм, даже опытные педиатры и невропатологи не всегда могут ее диагностировать. В патологический процесс вовлекается не только нервная система, но и соединительнотканные волокна. С этим связано поражение кожного покрова, связок, костей и суставов. Адекватная жизненная среда и специальные учебные программы позволяют большинству детей с СМБ учиться ходить, говорить, читать и писать.
Диагностика
Только квалифицированный генетик может правильно поставить диагноз на основании результатов специфических генетических тестов и анализов, выявляющих дефектную хромосому.

- Клинический метод — визуальный осмотр пациента, выслушивание жалоб, сбор анамнеза и обнаружение характерных клинических признаков. Этот метод недостаточно точен и информативен. По его результатам невозможно поставить окончательный диагноз.
- Цитогенетический метод является основным в диагностике недуга. У больных берут популяцию клеток и воздействуют на нее фолиевой кислотой, провоцирующей изменения хромосомы. Спустя некоторое время на ней обнаруживается область выраженного истончения. Это диагностический критерий синдрома. Цитогенетическое исследование дает точные результаты только на начальных стадиях развития патологии. По мере прогрессирования болезни оно теряет свою точность и специфичность, что связано с использование фолиевой кислоты и поливитаминов, содержащих ее.
- Кариотипирование – исследование набора хромосом. При обнаружении измененной хромосомы генетики ставят диагноз.
- Молекулярно-генетический анализ позволяет определить количество повторов нуклеотидов в гене.
- Полицепная реакция – высокоспецифичный метод, с помощью которого специалисты изучают структуру аминокислот в Х-хромосоме и определяют наличие СМБ.
- Электроэнцефалография позволяет определить схожую биоэлектрическую мозговую активность у больных с синдромом.
Пренатальная диагностика проводится с целью обнаружения патологии на стадии внутриутробного развития. Неинвазивные методы заключаются в ультразвуковом исследовании беременной женщины и скрининге сывороточных факторов крови матери. Инвазивные методы — исследование пуповинной крови, биопсия хориона, амниоцентез, плацентоцентез. Если во время исследования были обнаружены признаки синдрома, женщине предлагают сделать аборт или оставить беременность, но пройти специфическое внутриутробное лечение плода.
Лечение

Синдром Мартина-Белла – генетически детерминированное заболевание, которое полностью не излечивается. Общетерапевтические мероприятия направлены на устранение основных симптомов недуга и облегчение жизни больных. Комплексная терапия позволяет добиться максимально эффективных результатов.
Пациентам назначают следующие группы препаратов:
- антидепрессанты – «Кломипромин», «Флюоксегин», «Флювоксамин»,
- нейролептики – «Галоперидол», «Перициазин»,
- психостимуляторы – «Кортексин», «Кавинтон», «Фезам»,
- седативные средства – «Диазепам», «Седуксен»,
- ангиопротекторы или сосудистые препараты – «Церебролизин», «Винпоцетин», «Актовегин»,
- ноотропы – «Ноотропил», «Пирацетам»,
- средства, разжижающие кровь – «Плавикс», «Клексан», «Синкумар»,
- противоэпилептические средства – «Конвулекс», «Мазепин»,
- препараты лития – «Седалит», «Литарекс»,
- поливитаминные комплексы.
Кроме лекарственной терапии всем больным показаны физиотерапевтические процедуры – плавание, водная гимнастика, миорелаксация, иглоукалывание. Уменьшить проявления синдрома позволяют занятия с логопедами и педагогами. Опытные психотерапевты и психологи во время занятий помогут избавиться от сильного стеснения, постоянного страха, необдуманных поступков и навязчивых идеей.
Хирургическое лечение проводится при синдроме Мартина-Белла. Пластические операции по восстановлению формы ушей, конечностей и половых органов улучшают внешность больного. Хирурги корректируют гинекомастию и прочие внешние дефекты.
Народная медицина позволяет снять напряжение, тревогу и улучшить сон. Готовят отвары и настои из лекарственных трав – валерианы, мяты, календулы, пустырника, зверобоя, ромашки.
Эффективность лечения синдрома Мартина-Белла все еще остается не очень высокой, несмотря на разработки современных ученых-медиков. Все лечебные мероприятия оказывают лишь временный результат и поддерживают самочувствие больных на оптимальном уровне непродолжительный отрезок времени. При возобновлении клинических симптомов недуга врачи назначают повторный курс лечения. Многим пациентам активные терапевтические процедуры позволяют вести нормальный образ жизни. Правильно назначенное лечение тормозит дальнейшее развитие болезни.
Прогноз и рекомендации
Пренатальный скрининг беременных позволяет предупредить развитие синдрома. Если у одного из родителей имеется пораженная хромосома, показано экстракорпоральное оплодотворение, которое позволяет родить здорового ребенка.
Прогноз при синдроме Мартина-Белла считается относительно благоприятным, несмотря на то, что недуг неизлечим. Больные живут долго, но рано становятся инвалидами. Дети чаще болеют инфекционными заболеваниями и в наибольшей степени повержены травмам.
Специалисты рекомендуют родителям помнить, что их ребенок – это личность со своими правами, чувствами и потребностями, как и все остальные дети. Они нуждаются в любви и внимании своих родных и близких. Чтобы больной малыш чувствовал себя комфортно, необходимо спокойно и достойно реагировать на пристальные взгляды окружающих, без стеснения отвечать на вопросы друзей и близких. Главное понять, что не все вокруг могут одинаково искренне сопереживать и воспринимать «особенных» детей.
Видео: презентация по синдрому Мартина-Белла
Источник