
Синдром марфана этиология патогенез клиника диагностика лечение

Синдром Марфана: причины, диагностика, лечениеЭтиология и встречаемость синдрома Марфана. Синдром Марфана (MIM №154700) — панэтническое аутосомно-доминантное заболевание соединительной ткани, вызванное мутациями в гене фибриллина 1 (FBN1, MIM № 134797). Синдром Марфана имеет встречаемость около 1 на 10 000. Приблизительно 25-35% пациентов имеют новую мутацию. Мутации, вызывающие синдром Марфана, разбросаны по гену, и каждая мутация обычно уникальна в семье. Патогенез синдрома МарфанаГен FBN1 кодирует фибриллин 1, внеклеточный матричный гликопротеид с широким распределением. Фибриллин 1 полимеризуется, формируя микрофибриллы как в эластичных, так и в неэластичных тканях, например стенке аорты, цилиарных поясках и коже. Мутации влияют на синтез, процессинг, секрецию, полимеризацию или устойчивость фибриллина 1. Исследования накопления и экспрессии фибриллина 1 в культуре клеток предположили доминантный отрицательный патогенез, т.е. синтез мутантного фибриллина 1 тормозит образование нормальных микрофибрилл нормальным фибриллином 1 или стимулирует протеолиз несоответствующих внеклеточных микрофибрилл. Последние исследования на мышиных моделях синдрома Марфана указывают, что половинного количества нормального фибриллина 1 недостаточно, чтобы проводить эффективную сборку микрофибрилл. Таким образом, патогенезу болезни также может содействовать гаплонедостаточность. Кроме синдрома Марфана, мутации в гене FBN1 могут вызывать другие синдромы, включая неонатальный синдром Марфана, изолированные скелетные симптомы, аутосомно-доминантную эктопию хрусталиков и фенотип MASS (марфаноидные симптомы, включая пролапс митрального клапана или миопию, пограничное и непрогресирующее расширение аорты, и неспецифические изменения скелета и кожи). В общем, фенотипы довольно схожи в пределах семьи, хотя тяжесть фенотипических проявлений может значительно изменяться. До настоящего времени точное соотношение между генотипом и фенотипом не определено. Внутрисемейная и межсемейная изменчивость позволяет предполагать, что в определении фенотипа важную роль играют окружающая среда и эпигенетические факторы. Последние исследования на мышиных моделях показывают, что фибриллин 1 не просто структурный белок, и что синдром Марфана не просто результат структурной слабости тканей. Более того, микрофибриллы фибриллина 1 в норме связывают и уменьшают концентрацию и активность факторов роста суперсемейства TGFb. Потеря фибриллина 1 увеличивает сигналы свободного TGFb значительно содействующие заболеванию, так как антагонисты TGFb устраняют легочные и клапанные изменения, наблюдаемые у мышей с недостаточностью фибриллина 1.
Фенотип и развитие синдрома МарфанаСиндром Марфана — мультисистемное заболевание со скелетными, глазными, сердечно-сосудистыми, легочными, кожными и другими аномалиями. Скелетные аномалии включают очень высокий рост (отношение размаха рук к росту >1,05; соотношение верхнего и нижнего сегментов <0,85 у взрослых), арахнодактилию, аномалии грудины, сколиоз, разболтанность суставов, готическое нёбо. Аномалии глаз включают подвывих хрусталиков, уплощение роговицы, удлинение глазного яблока и гипоплазию радужки. Сердечнососудистые аномалии включают пролапс митрального клапана, аортальную регургитацию и расширение и расслаивающую аневризму восходящей аорты. Легочные аномалии включают спонтанный пневмоторакс и расширение концевых пузырьков. Аномалии кожи включают атрофические бороздки и рецидивирующие грыжи. Аномалии твердой мозговой оболочки включают выбухание оболочки в крестцово-поясничном отделе. Большинство признаков синдрома Марфана появляются с возрастом. Скелетные аномалии типа аномалии грудины и сколиоза ухудшаются с ростом костей. Подвывих хрусталика часто присутствует уже в раннем детстве, но может развиваться и в юности. С повышенной частотой при синдроме Марфана встречаются отслойка сетчатки, глаукома и катаракты. Сердечно-сосудистые осложнения обнаруживаются в любом возрасте и развиваются в течение всей жизни. Основные причины преждевременной смерти пациентов с синдромом Марфана — сердечная недостаточность вследствие регургитации клапанов и аневризмы и разрыва аорты. Тем не менее в связи с улучшением хирургической и терапевтической помощи при аневризме аорты выживание улучшилось. С 1972 по 1993 г. ожидаемый возраст выживания для 50% пациентов поднялся с 49 до 74 лет для женщин и с 41 до 70 лет для мужчин. Особенности фенотипических проявлений синдрома Марфана:
Лечение синдрома МарфанаСиндром Марфана — клинический диагноз, определяемый по наличию конкретных симптомов. Подтверждение синдрома Марфана идентификацией мутаций в гене FBN1 в настоящее время практически нецелесообразно, поскольку крайняя аллельная гетерогенность делает идентификацию причинно-обусловленной мутации в каждой семье крайне трудозатратной, а также из-за недостаточно надежной корреляции между генотипом и фенотипом. Анализ мутаций имеет недостаточную чувствительность или специфичность для синдрома Марфана, что ограничивает его клиническую пользу. Для синдрома Марфана недоступно эффективное лечение; следовательно, помощь сфокусирована на профилактике осложнений и симптоматическом лечении. Оказание офтальмологической помощи включает регулярные осмотры, коррекцию миопии и, часто, замену хрусталика. Ортопедическая помощь заключается в укрепляющем лечении или хирургической коррекции сколиоза. Помощь при аномалии грудины в основном косметическая. Физиотерапия может компенсировать нестабильность суставов. Сердечно-сосудистые проблемы решаются комбинацией терапевтических и хирургических мероприятий. Терапевтические усилия направлены на предохранение или замедление развития расширения корня аорты за счет уменьшения кардиологических показателей, снижения артериального давления и усилия выброса желудочков с помощью бета-адреноблокаторов, ограничение участия в контактных видах спорта, соревновательных видах спорта и в изометрических упражнениях. Профилактическая замена корня аорты показана, когда расширение аорты или аортальная регургитация становится достаточно тяжелой. Большинству пациентов в настоящее время проводят надклапанную замену корня аорты, не требующую постоянного приема противосвертывающих препаратов. Гемодинамические изменения, связанные с беременностью, могут приводить к прогрессирующему расширению или расслоению аорты. Полагают, что расслоение аорты вторично к гормональным изменениям, увеличению объема крови и сердечного выброса, связанных с беременностью и родами. Современные исследования считают, что риск беременности слишком велик, если ширина корня аорты превышает 4 см. Женщины могут выбрать проведение надклапанной замены аорты перед беременностью. Риски наследования синдрома МарфанаПациенты с синдромом Марфана имеют 50% риск иметь ребенка, больного синдромом Марфана. В семьях, передающих синдром Марфана, членов семьи, находящихся в группе риска, можно выявлять, либо обнаруживая мутацию (в тех редких случаях, когда она известна), либо анализом сцепления, если маркеры, тесно сцепленные с локусом FBN1, имеют очевидную связь с болезнью в семье пробанда. Пренатальная диагностика доступна только для семей, в которых возможны исследования сцепления или известна мутация в гене FBN1. Пример синдрома Марфана. Д.Л., здоровый 16-летний ученик средней школы, звезда баскетбола, направлен в генетическую клинику для обследования по поводу синдрома Марфана. Телосложение Д.Л. похоже на телосложение его отца. Отец Д.Л., высокий субтильный человек, умер во время утренней пробежки; у других членов семьи случаев скелетных аномалий, внезапной смерти, снижения зрения или врожденных аномалий не было. При медицинском осмотре выявлены астеническое телосложение, высокое дугообразное нёбо, небольшая деформация грудины по типу «куриной» груди, арахнодактилия, соотношение размах рук/рост 1,1, диастолический шум и стрии на плечах и бедрах. Эхокардиография выявила расширение корня аорты с аортальной регургитацией. Офтальмологическое обследование показало двусторонний иридодонез и легкое смещение хрусталиков кверху. На основе медицинского осмотра и результатов обследования генетик объяснил пациенту и его матери, что у него синдром Марфана. – Также рекомендуем “Синдром Миллера-Дикера: причины, диагностика, лечение” Оглавление темы “Наследственные болезни”:
|
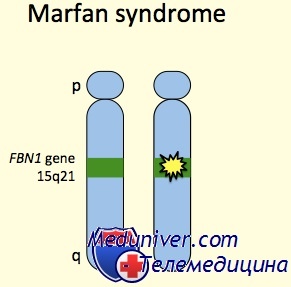
Источник
Что такое Болезнь (синдром) Марфана –
Болезнь Марфана (синдром Марфана) – это врожденная коллагенопатия или системная недостаточность соединительной ткани, для которой характерный определенный тип наследования и выраженной клинической симптоматики: проявления скелетной, сердечно-сосудистой и глазной патологии. Больные с синдромом Марфана имеют в своем анамнезе аневризмы аорты, гигантизм, миопию, долихостеномелию и арахнодактилию, деформацию грудной клетки, протрузию вертлужной впадины, эктопию хрусталика, плоскостопие, эктазию твердой мозговой оболочки, кифосколиоз.
Заболевание получило название в честь французского педиатра Антуана Марфана, который впервые в 1886 г. описал пятилетнюю девочку, имеющую длинные тонкие ноги и «паучьи» пальцы.
Соотношение распространенности данного врожденного заболевания составляет примерно 1:10 000 людей и мало зависит от пола. Однако процентный показатель среди детей и подростков достигает 6,8%, отметим, что большую часть из них формируют мальчики.
Что провоцирует / Причины Болезни (синдрома) Марфана:
Синдром Марфана – это врожденная аномалия, наследуемая по аутосомно-доминантному типу. Причиной возникновения синдрома являются мутации гена FBN1, который отвечает за синтез фибриллина – структурного белка межклеточного матрикса, предоставляющего эластичность и сократимость соединительной ткани. Аномалия и нехватка фибриллина провоцирует нарушения формирования волокнистых структур и потерю прочности и упругости соединительной ткани, невозможность выдержать физиологические нагрузки. В стенках сосудов эластического типа происходят гистологические изменения. В 75 % случаев синдром Марфана имеет семейный тип наследования, а в 25 % происходит первичная мутация. Если отцу более 35 лет и в его анамнезе есть синдром Марфана, то очень высокая вероятность того, что он передаст заболевание ребенку.
Патогенез (что происходит?) во время Болезни (синдрома) Марфана:
Ученые выделяют несколько форм синдрома Марфана, которые зависят от количества пораженных органов и систем:
- стертая – имеет слабо выраженные изменения в 1-2-х системах;
- выраженная – имеет слабо выраженные изменениями в 3-х системах; или выраженные изменения хотя бы в 1-ой системе; или выраженные изменения в 2-3-х и более системах.
При синдроме Марфана степень тяжести изменений разделяется на легкую, среднюю и тяжелую. По характеру течения выделяют прогрессирующий и стабильный синдром Марфана.
Симптомы Болезни (синдрома) Марфана:
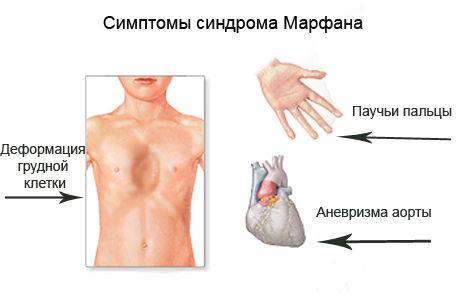
Клинические симптомы заболевания разделяют на несколько групп, которые отражают точную локализацию проявлений соединительнотканной (мезенхимальной) дисплазии:
- костно-суставные расстройства (астеническая конституция, узкий лицевой череп с «птичьим» выражением лица, плоскостопие, узкая и деформированная, килевидная или воронкообразная грудная клетка, арахнодактилия кистей и стоп, кифосколиотическая деформация позвоночника, гипермобильность суставов и сухожилий). Костно-суставные нарушения есть у большинства больных;
- изменения мягких тканей (малая масса тела, мышечная гипотония, гипоплазия жировой ткани и мускулатуры, плоскостопие);
- изменения внутренних органов (аневризма восходящей аорты, клапанные пролапсы, особенно пролапс митрального клапана, гипоплазированное, расширение корня аорты и легочной артерии, аневризмы синусов Вальсальвы, «висячее», «капельное» сердце, уменьшение долей легких, слишком длинный и гипопластичный кишечник);
- нарушения в системе зрения (голубые склеры, гиперметропия высоких степеней, аниридия, выраженная миопия, эктопия и подвывихи хрусталика, афакия, колобома);
- расстройства центральной нервной системы (анизокория, нистагм, асимметрия сухожильных рефлексов, пирамидные расстройства);
- Расстройства гипофизарно-адреналовой системы (высокий рост, акромегалоидные расстройства, несахарный диабет, арахнодактилия, удлиненные конечности, увеличенные ступни, вегетативные расстройства);
- нарушения сердечно-сосудистой системы (нарушения внутрижелудочковой проводимости, умеренные признаки гипертрофии миокарда левого желудочка и предсердия, изменения в сердце и магистральных сосудах, аортальная недостаточность, пролапс митрального клапана, нарушения внутрисердечной гемодинамики, расширение корня аорты, двустворчатый клапан аорты, митральная недостаточность, которая связана с развитием миксоматозной дегенерацией створок, увеличением их площади, расширением фиброзного кольца, удлинением хорд, «разболтанностью» створок и увеличением пролабирования). У больных наблюдается мышечная слабость и снижение активности при физических нагрузках.
Диагностика Болезни (синдрома) Марфана:
Диагноз синдрома Марфана ставится на основе семейного анамнеза, выраженности симптоматики, физикального осмотра, результатов функционального, рентгенологического, офтальмологического и генетического исследований, ЭКГ и ЭхоКГ и лабораторных исследований.
Во время диагностирования учитываются фенотипические диагностические тесты, которые определяют соотношение кисти и роста, длину среднего пальца, индекс телосложения Варги, тест большого пальца на арахнодактилию, тест охвата запястья.
ЭКГ определяет нарушения ритма сердца, выраженную гипертрофию миокарда.
ЭхоКГ обнаруживает дилатацию аорты, клапанную регургитацию, пролапс митрального клапана, увеличение размеров левого желудочка, разрывы хорд.
Рентгенография грудной клетки позволяет увидеть расширение дуги аорты и корня, увеличение размеров сердца; рентгенография тазобедренных суставов показывает протрузию вертлужной впадины
КТ и МРТ сердца и сосудов делают для того, чтобы выявить дилатацию и аневризмы аорты; МРТ позвоночника показывает эктазию твердой мозговой оболочки.
Аортография проводится при подозрении на аневризму и расслоение аорты.
Биомикроскопия и офтальмоскопия показывают эктопию хрусталика.
Генетическая идентификация показывает изменения (мутации) в гене FBN1.
При синдроме Марфана прибегают к дифференциальной диагностике с болезнями, схожими с синдромом, к ним относятся: гомоцистинурия, врожденная контрактурная арахнодактилия (синдром Билса), наследственная артроофтальмопатия (синдром Стиклера), MASS-синдром, синдром Элерса-Данлоса, Лойса-Дитца, Шпринцена-Голдберга, семейная эктопия хрусталика и др.
Лечение Болезни (синдрома) Марфана:
К терапии синдрома Марфана относится консервативная и хирургическая коррекция сердечно-сосудистых нарушений, поражений скелета и органы зрения.
Лечение, прежде всего, направлено на профилактику развития заболевания и осложнений в сердечно-сосудистой системе. Если диаметр аорты до 4 см больному назначают антагонисты кальция, β-адреноблокаторы или ингибиторы АПФ. Хирургическое вмешательство проводится только, когда диаметр аорты составляет больше 5 см при пролапсе митрального клапана, недостаточности клапанов сердца, восходящей части и расслоении аорты.
При синдроме Марфана выполняют реконструктивные операции на аорте. В случае необходимости выполняется протезирование митрального клапана.
Больным с синдромом Марфана назначается коррекция зрения методом подбора очков и контактных линз, а в сложных случаях – путем лазерного или хирургического лечения болезней зрения.
Детям со скелетными нарушениями проводят хирургическую стабилизацию позвоночника, эндопротезирование тазобедренных суставов, торакопластику.
В курс лечения входит: витаминотерапия, патогенетическая коллагеннормализующая и метаболическая терапия.
Профилактика Болезни (синдрома) Марфана:
Больные с данным синдромом находятся под постоянным наблюдением врачей, они регулярно проходят диагностическое обследование. Детям разрешена легкая физическая нагрузка, исключающая спортивные соревнования, подводное плавание, контактные виды спорта.
К профилатике относится своевременная кардиохирургическая коррекция, которая увеличивает продолжительность жизни до 70 лет.
К каким докторам следует обращаться если у Вас Болезнь (синдром) Марфана:
Кардиолог
Офтальмолог
Кардиохирург
Ортопед
Генетик
Терапевт
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Болезни (синдрома) Марфана, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу.
Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас ? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни. Определение симптомов – первый шаг в диагностике заболеваний в целом. Для этого просто необходимо по несколько раз в год проходить обследование у врача, чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой. Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина. Также зарегистрируйтесь на медицинском портале Eurolab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Другие заболевания из группы Болезни ребенка (педиатрия):
Если Вас интересуют еще какие-нибудь виды болезней и группы заболеваний человека или у Вас есть какие-либо другие вопросы и предложения – напишите нам, мы обязательно постараемся Вам помочь.
Источник