
Синдром меккеля что это такое

Синдром Меккеля. Диагностика и прогноз при синдроме Меккеля
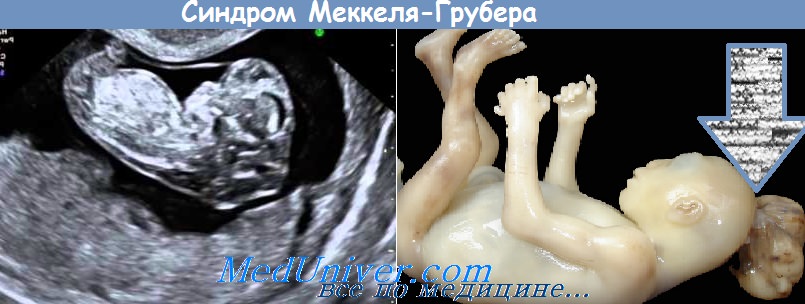
Это редко встречающийся летальный синдром, который характеризуется затылочным цефалоцеле, постаксиальной полидактилией и кистозной дисплазией почек. Он может сочетаться со многими другими состояниями, наиболее распространенным из которых является фиброз печени.
Синонимы. Дисэнцефалия с кистозными изменениями внутренних органов и синдром Меккеля (Meckel) (принят в англоязычной литературе). Синдром Меккеля (Meckel) является более предпочтительным названием заболевания и используется как при поиске в интернет-базе данных Medline, так и в Британской энциклопедии врожденных пороков (Birth Defect Encyclopedia). Другими названиями являются синдром Грубера (Gruber) (используется в европейской литературе) и синдром Меккеля-Грубера (Meckel-Gruber).
Распространенность. Точно неизвестна, однако по общепринятому мнению это очень редко встречающаяся патология. По данным D. Bergsma, распространенность синдрома Меккеля (Meckel) составляет 0,2 на 10 000 новорожденных. R. Salonen и R. Norio установили, что частота данного синдрома при рождении варьирует от 0,07 до 0,7 на 10 000 родов.
Генетические нарушения. Локус, ответственный за возникновение синдрома Меккеля (Meckel), локализован на длинном плече хромосомы 17q2.1-q2.4. Фенотипическая вариабельность и случаи без подтвержденной связи с хромосомой 17q предполагают наличие некоторой степени локусной гетерогенности.
Диагностика. В 1981 году F.C. Fraser и A. Lytwyn предположили, что кистозная дисплазия почек является постоянной аномалией при синдроме Меккеля (Meckel), и поэтому для установления диагноза она должна обнаруживаться в сочетании с не менее чем двумя меньшими дефектами. Данная концепция продолжает обсуждаться, при этом распространенность аномалий почек при данном синдроме составляет от 95 до 100%. Почки первоначально имеют микроскопические кисты, которые развиваются, разрушая ее паренхиму и вызывая увеличение органа в 10-20 раз.
Первым эхографическим признаком в большинстве случаев является маловодие вследствие дисфункции почек, возникающее в начале второго триместра беременности, когда продукция мочи должна замещать внеклеточную диффузию в качестве основного источника околоплодных вод. Однако в некоторых случаях синдрома может обнаруживаться нормальное количество амниотической жидкости, и, таким образом, ее наличие не исключает диагноза. В ряде ситуаций мочевой пузырь не будет визуализироваться. Отсутствие эхографических признаков заболевания на ранних сроках у беременной из группы риска по рецидиву заболевания у плода не исключает синдрома Меккеля (Meckel), и в этих случаях рекомендуется наблюдение в динамике до 20 нед гестации.
Затылочное цефалоцеле встречается в 60-80%. В связи с тем, что цефалоцеле окружено мембраной, показатели уровня АФП в крови матери или в амниотической жидкости могут быть нормальными. Постаксиальная полидактилия встречается в 55-75%. Также могут присутствовать другие аномалии конечностей, в виде их искривления и укорочения. Выявление, по крайней мере, двух из трех признаков классической триады при наличии нормального кариотипа позволяет установить диагноз.
Дифференциальный диагноз. Дифференциальный диагноз будет зависеть от типа сочетанных аномалий. Вследствие сходства нескольких эхографических признаков трисомию 13 следует исключить путем кариотипирования. Другим возможным заболеванием является аутосомно-доминантная поликистозная болезнь почек.
Сочетанные аномалии. Перечень возможных аномалий, сочетающихся с данным синдромом, является обширным. В некоторых ситуациях такая широкая фенотипическая вариабельность значительно затрудняет установление диагноза.
Прогноз. Синдром Меккеля (Meckel) является летальным заболеванием. В большинстве случаев отмечаются мертворождения или ранняя неонатальная гибель в течение нескольких часов или дней после рождения. В некоторых наблюдениях описано выживание в течение нескольких месяцев, но с низким качеством жизни. Н.М. Ramadani и Н.А. Nasrat сообщили о ребенке, который умер в возрасте 28 месяцев. В 1997 году P. Paavola et al. привели наблюдение еще одного нетипичного случая выживания в течение 18 месяцев.
Акушерская тактика. При подозрении на синдром Меккеля (Meckel) необходимо провести кариотипирование с целью исключения хромосомных заболеваний. Если принимается решение о ее пролонгировании или если диагноз установлен в более поздние сроки, стандартная акушерская тактика ведения беременной не изменяется.
– Также рекомендуем “Мезомелическая дисплазия. Диагностика и прогноз мезомелической дисплазии”
Оглавление темы “Врожденные синдромы плода”:
1. Синдром Клиппеля-Треноне-Вебера. Синдром Ларсена у плода
2. Синдром летального птеригума. Лиссэнцефалия I типа
3. Синдром Меккеля. Диагностика и прогноз при синдроме Меккеля
4. Мезомелическая дисплазия. Диагностика и прогноз мезомелической дисплазии
5. Микроцефалический первичный нанизм. Синдром Неу-Лаксова
6. Синдром Нунан у плода. Секвенция маловодия
7. Несовершенный остеогенез. Диагностика и прогноз несовершенного остеогенеза у плода
8. Синдром Пена-Шокейра. Пентада Кантрелла
9. Синдром Пфайффера. Диагностика и прогноз синдрома Пфайффера
10. Синдром Поланда. Секвенция Поттер и синдром prune-belly
Источник
Что такое синдром Меккеля?
Синдром Меккеля — это редкое наследственное заболевание, характеризующееся аномалиями, затрагивающими несколько систем организма. Больные с синдромом Меккеля обычно испытывают три классических симптома: выпячивание части головного мозга и окружающих его оболочек (менингиев) через дефект в задней части черепа (энцефалоцеле), множественные кисты в почках (кистозные поражения почек) и дополнительные пальцы рук и ног (полидактилия). Пострадавшие дети или плоды также могут иметь нарушения, затрагивающие голову и лицо (черепно-лицевую область), печень, легкие, сердце и мочеполовые пути. Недостаток околоплодных вод, окружающих плод (маловодие), вызывает неполное развитие легких (легочная гипоплазия).
Из-за этих серьезных проблем со здоровьем дети, рожденные с синдромом Меккеля, не живут дольше, чем несколько дней или недель. Большинство пострадавших младенцев умирают от почечной недостаточности или респираторных заболеваний. Родители иногда решают прервать беременность, когда у плода обнаруживается синдром Меккеля во время беременности.
Синдром Меккеля наследуется как аутосомно-рецессивное состояние через тринадцать генов: B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRIP1L, TCTN2, TCTN3, TMEM67, TMEM107, TMEM216, TMEM231 и TMEM237.
Первое сообщение о синдроме Меккеля было опубликовано Иоганном Фридрихом Меккелем в 1822 году. В 1934 году Г.Б. Грубер опубликовал отчеты о людях с синдромом Меккеля и назвал расстройство спланхнокистозная дизэнцефалия. Часто расстройство также называют синдромом Меккеля — Грубера.
Признаки и симптомы

Специфические симптомы, связанные с синдромом Меккеля, сильно различаются у разных людей. У пострадавших детей не будет всех симптомов, описанных ниже. Нарушения центральной нервной системы, легких или почек всегда приводят к перинатальной смерти.
Наиболее распространенной аномалией центральной нервной системы, связанной с синдромом Меккеля, является затылочный энцефалоцеле, состояние, при котором ребенок рождается с разрывом в черепе (т.е. часть одной или нескольких пластин, образующих череп, не герметизируется). Мембраны, которые покрывают мозг (мозговые оболочки) и ткани мозга, часто выступают через этот разрыв. Затылочный энцефалоцеле может привести к накоплению избыточного количества спинномозговой жидкости (СМЖ) в черепе, что вызывает давление на ткани головного мозга (гидроцефалия). Дополнительные нарушения центральной нервной системы, которые могут возникать у детей с синдромом Меккеля, включают в себя отсутствие большей части головного мозга, черепа и кожи головы (анэнцефалия), отсутствие срединной части заднего мозга (агенезия червя мозжечка) и состояние, известное как микроцефалия, при котором окружность головы меньше, чем можно было бы ожидать для возраста и пола ребенка.
У пострадавших детей могут быть характерные черты лица, включая:
- аномально маленькую челюсть (микрогнатия);
- увеличенные, низко посаженные и уродливые уши;
- волчья пасть;
- заячья губа;
- наклонный лоб;
- короткая шея.
У пострадавших детей могут быть глазные аномалии, включая аномально маленькие глаза (микрофтальмия) и недоразвитие нервов глаз (гипоплазия зрительного нерва или колобома). Множественные кисты в почках (мультикистозная дисплазия почек) являются наиболее распространенным симптомом, связанным с синдромом Меккеля. Состояние характеризуется нормальной почечной тканью, которая заменяется заполненными жидкостью мешочками или кистами различных размеров, которые становятся больше (в 10-20 раз больше, чем обычно) по мере прогрессирования заболевания. Симптомы, связанные с кистозными поражениями почек, включают потерю функции почек, что приводит к терминальной стадии почечной недостаточности.

Пострадавшие могут также иметь лишние пальцы рук и ног (полидактилия). Дополнительные пороки развития скелета включают в себя изгиб длинных костей рук и ног, искривление пятого пальца (клинодактилия), сращивании пальцев кисти/стопы (синдактилия) и косолапость.
У некоторых людей могут присутствовать аномалии мочеполового тракта, включая неспособность одного или обоих яичек опускаться в мошонку (крипторхизм), недоразвитый мочевой пузырь и неполное развитие гениталий.
У некоторых пострадавших детей могут быть нарушения, затрагивающие другие органы тела, включая печень, легкие или сердце. В печени может появится фиброзная ткань (фиброз печени) и расширение (дилатация) и чрезмерное количество небольших проходов, которые переносят желчь из печени в тонкую кишку (желчные протоки). Легкие могут быть недоразвиты, а структура, которая покрывает вход в гортань при глотании, может быть раскрыта (расщелина надгортанника). Селезенка может отсутствовать (аспления) или присутствовать в виде множественных маленьких селезенок, а не одной (полиспления).
Сердечные аномалии могут включать дефекты межпредсердной и межжелудочковой перегородки (ДМПП и ДМЖП) и открытый артериальный проток или другие более сложные пороки развития. ДМПП характеризуются аномальным отверстием в фиброзной перегородке, которая разделяет две верхние камеры (предсердия) сердца. ДМЖП характеризуются аномальным отверстием в перегородке, которое разделяет две нижние камеры сердца (желудочки). Размер, расположение и характер дефекта перегородки и любые связанные с этим нарушения определяют степень выраженности симптомов. Открытый артериальный проток — состояние, при котором канал (проток) между кровеносным сосудом, который ведет к легким (легочная артерия) и главной артерией тела (аорта), не может закрыться после рождения.
Причины
Синдром Меккеля может быть вызван изменениями (мутациями) в тринадцати генах: B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRIP1L, TCTN2, TCTN3, TMEM67, TMEM107, TMEM216, TMEM231 и TMEM237 . Мутации в этих 13 генах составляют 75 процентов всех случаев; остальные 25 процентов имеют неизвестные генетические причины. Большинство из этих генов также ответственны за неврологическое расстройство, называемое синдромом Жубера, что приводит к концепции, что синдром Меккеля является крайней летальной формой синдрома Жубера.
Известно, что белки, продуцируемые этими генами, влияют на клеточные структуры или функции, называемые первичными ресничками. Реснички — это микроскопические проекции, которые торчат на поверхности клетки и помогают передавать информацию по сигнальным путям. Реснички важны для многих функций клеток, во многих типах клеток, особенно в почках, печени, глазах и головном мозге. Мутации в этих генах вызывают проблемы в функции первичных ресничек, что приводит к различным дефектам, зависящим от типа клеток. Ранняя дефектная цилиарная функция может быть причиной аномалий развития, особенно в почках, мозге, конечностях, сердце.
Синдром Меккеля наследуется как аутосомно-рецессивное генетическое заболевание. Рецессивные генетические нарушения возникают, когда человек наследует один и тот же измененный ген по одному признаку от каждого родителя. Если человек получает один рабочий ген и один нерабочий ген заболевания, то человек будет носителем заболевания, но обычно бессимптомным. Риск того, что оба родителя-носителя передадут нерабочий ген и, следовательно, заведут больного ребенка, составляет 25% при каждой беременности. Риск рождения ребенка-носителя, как и родители, составляет 50% с каждой беременностью. Вероятность для ребенка получить нормальные гены от обоих родителей составляет 25%. Риск одинаков для мужчин и женщин.
Родители, которые являются близкими родственниками (брат и сестра по крови), имеют больше шансов, чем несвязанные родители, иметь один и тот же ненормальный ген, что повышает риск рождения ребенка с рецессивным генетическим расстройством.
Затронутые группы населения
Синдром Меккеля поражает мужчин и женщин в равных количествах. В медицинской литературе зарегистрировано более 200 случаев. По оценкам, заболеваемость синдромом Меккеля в различных регионах мира составляет от 1 на 13 250 до 1 на 140 000 живорождений. Расстройство чаще встречается в финском населении из-за эффекта основателя, с частотой 1 на 9000 и 1 на 3000 лиц бельгийского происхождения. Тем не менее, у гуджаратцов болезнь имеет распространенность 1 на 1300. Расстройство происходит чаще в контексте родственных союзов.
Близкие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Меккеля. Сравнения могут быть полезны для дифференциальной диагностики:
- Синдром Смита-Лемли-Опица (дефицит 7-дегидрохолестерол редуктазы) представляет собой редкое наследственное расстройство развития, характеризующееся множественными врожденными нарушениями. Симптомы могут включать характерные черты лица, микроцефалию, маленький рост, умственную отсталость, лишние пальцы рук или ног (полидактилия), потерю зрения, неполное развитие мужских половых органов, короткий нос со смещенными ноздрями и стеноз привратника. У некоторых пациентов могут присутствовать аномалии мозга или сердца. Специфические симптомы, связанные с каждым человеком, сильно различаются. Синдром Смита-Лемли-Опица наследуется по аутосомно-рецессивному типу.
- Трисомия 13 (синдром Патау) представляет собой хромосомное расстройство, при котором вся или часть хромосомы 13 дублируется три раза (трисомия), а не дважды в клетках организма. У некоторых затронутых людей только процент клеток может содержать дополнительную 13-ю хромосому (мозаицизм), тогда как другие клетки содержат нормальную хромосомную пару. У людей с трисомией 13 диапазон и степень выраженности ассоциированных симптомов и признаков могут зависеть от конкретного расположения дублированной (трисомной) части хромосомы 13, а также от процента клеток, содержащих аномалии. Тем не менее, у многих пострадавших младенцев и детей такие отклонения могут включать задержки развития, глубокую умственную отсталость, необычно маленькие глаза (микрофтальмия), ненормальное углубление в верхней губе (заячья губа), неполное закрытие крыши рта (расщелина нёба), неопущенные яички (крипторхизм) у мужчин и лишние пальцы рук и ног (полидактилия). Могут также присутствовать дополнительные пороки развития черепно-лицевой области, такие как относительно небольшая головка (микроцефалия); широкий плоский нос; широко расставленные глаза; вертикальные кожные складки, закрывающие глаза; внутренние углы (эпикантальные складки); дефекты кожи головы; и уродливые, низко посаженные уши. У пострадавших детей может также быть неполное развитие определенных областей мозга (например, переднего мозга); почечные аномалии; врожденные дефекты сердца. Опасные для жизни осложнения могут развиться в младенчестве или в раннем детстве.
- Синдром коротких рёбер — полидактилии представляют собой группу редких скелетных нарушений, характеризующихся дефицитом роста, приводящим к невысокому росту, узкой грудной клетке и аномально коротким ребрам, а также дополнительным пальцам рук и ног. Существует значительное совпадение симптомов, связанных с синдромом коротких рёбер — полидактилии. Дополнительные признаки могут включать поликистоз почек, недоразвитие (гипоплазию) легких, аномалии мочеполовой системы, аномалии центральной нервной системы, задержки развития, расщелину губы и расщелину неба. Тяжелые формы синдрома коротких рёбер — полидактилии включают синдром Салдино-Нунана, Маевского, Верма-Наумова и Бимера-Лангера. Эти нарушения наследуются по аутосомно-рецессивному типу.
Диагностика
Диагноз синдрома Меккеля часто ставится на УЗИ во время беременности или при родах, при тщательном клиническом обследовании. Для подтверждения диагноза и проведения генетической консультации можно использовать молекулярно-генетическое тестирование. Пренатальная диагностика доступна с помощью ультрасонографии уже на 14 неделе беременности, которая может обнаружить определенные аномалии (например, энцефалоцеле, полидактилию, кистозные поражения почек и маловодие). Для исключения трисомии 13 может быть проведен хромосомный анализ. Для исключения синдрома Смита Лемли-Опица можно провести биохимическое исследование.
Стандартные методы лечения
В настоящее время нет никакого метода лечения синдрома Меккеля. Расстройство имеет постоянно летальный исход из-за почечной недостаточности и гипоплазии легких. Лечение симптоматическое и поддерживающее. Для семей рекомендуется генетическое консультирование.
Прогноз
Расстройство является летальным в утробе матери или в очень раннем неонатальном периоде с легочной гипоплазией и почечной недостаточностью в качестве основных причин ранней смерти.
Источник

SonoAce-R7
Универсальный ультразвуковой сканер высокого класса, ультракомпактный дизайн и инновационные возможности.
Синдром Меккеля-Грубера (дизэнцефалия спланхнокистозная) – комплекс множественных летальных врожденных пороков развития с аутосомнорецессивным типом наследования. Картировано несколько локусов синдрома: тип 1 (ген MKS1 [249000 – регистрационный номер по каталогу моногенных заболеваний OMIM] на хромосоме 17q23), тип 2 (ген MKS2 [603194] на 11q13), тип 3 (ген MKS3 [607361] на 8q), тип 4 (ген MKS4 [611134] на 12q21.3) [1].
Опорными признаками синдрома (“ядром”) являются: затылочная черепномозговая грыжа (80%), полидактилия (75%), обычно постаксиальная (чаще поражаются кисти), а также дисплазия почек (95%) [2]. В 15% случаев полидактилия сочетается с синдактилией. Могут наблюдаться камптодактилия, клинодактилия V, косолапость и поперечная ладонная складка.
Для диагностики синдрома, по мнению Г.И. Лазюка и соавт., достаточно наличия любых двух опорных признаков [3]. Кроме ключевых признаков синдрома, возможен широкий спектр сочетанных аномалий: микроцефалия (32%), гидроцефалия (25%), гипо- или аплазия мозолистого тела (13%), аринэнцефалия (17%), гипо- или аплазия мозжечка (28%), в единичных случаях анэнцефалия, гидроцефалия, голопрозэнцефалия, мальформация Dandy – Walker, септальные дефекты сердца, омфалоцеле, кистофиброз печени, расщелины лица и позвоночника. Встречаются также скошенный лоб, низко расположенные деформированные ушные раковины, микрогения, гипертелоризм, реже – гипотелоризм, капиллярные гемангиомы на лбу, зубы новорожденного, липоматозные образования на боковых поверхностях языка. В 25% случаев имеются пороки развития глаз: микрофтальмия (19,2%), реже – анофтальмия, колобомы, катаракта [2, 4].
Частота синдрома Меккеля-Грубера точно неизвестна. По данным B. Auber и соавт. [5], распространенность синдрома в Германии оценивается как 1:135000, в Израиле 1:50000, в Минске – 1:65000 родов [6]. В Финляндии и Татарстане это заболевание встречается необычно часто и достигает 1,1 на 10000 родов. Имеются данные, что оно составляет 5% всех дефектов нервной трубки [7].
В современной зарубежной периодике и в интернете опубликовано значительное количество сообщений об успешной пренатальной диагностике синдрома Меккеля-Грубера во II триместре беременности, публикации о более раннем распознавании этого синдрома единичны [4, 6, 8-15].
В связи с большой редкостью этого синдрома, единичными публикациями в отечественной литературе [6], посвященными его диагностике, представляем собственный опыт пренатальной диагностики 3 случаев синдрома Меккеля-Грубера, каждый из которых был интересен и уникален в плане постановки диагноза.
Наблюдение 1
Беременная А., 32 лет. Данная беременность 3-я, в анамнезе 1 неосложненные роды в срок и 1 артифициальный аборт. Наследственность не отягощена. Мужу 36 лет, здоров. Брак не родственный, супруги производственных вредностей не имеют. Пациентка при сроке гестации 13 недель 3 дня обратилась для проведения скрининговой эхографии в городской перинатальный центр (Ростов-на-Дону). Биохимический скрининг I триместра не проводился. Исследование выполнялось на ультразвуковом сканере с использованием режима поверхностной объемной реконструкции 3D/4D.
При эхографии в срок 13 недель 3 дня фетометрические параметры плода соответствовали гестационной норме, толщина воротникового пространства составила 1,5 мм; длина костей носа – по 2,5 мм. При трансвагинальном сканировании были обнаружены: затылочное энцефалоцеле размерами 7,4х3,5х3,7 мм (рис. 1), полидактилия кистей (рис. 2), увеличенные кистозно-измененные почки: правая – 28,5х21,3 мм; левая – 26,4х18,7 мм (рис. 3). Объем амниотической полости был нормальным для данного срока.
Рис. 1. Затылочное энцефалоцеле.
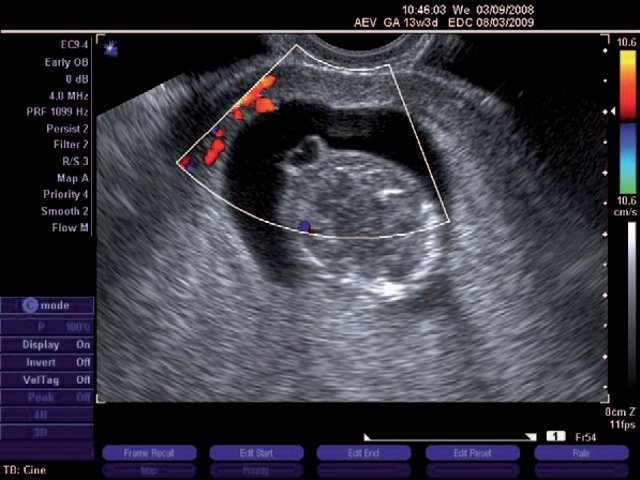
б) Режим ЦДК.
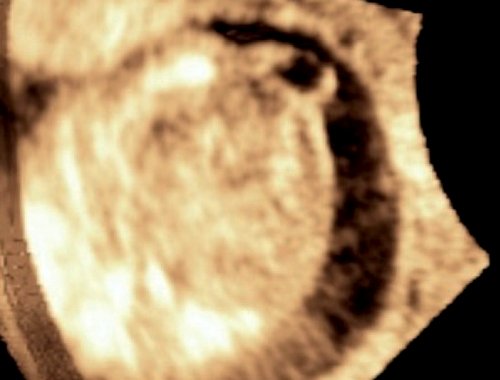
в) Режим 3D.
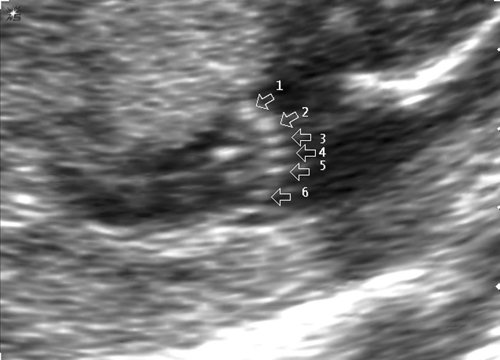
Рис. 2. Правая кисть плода, полидактилия.
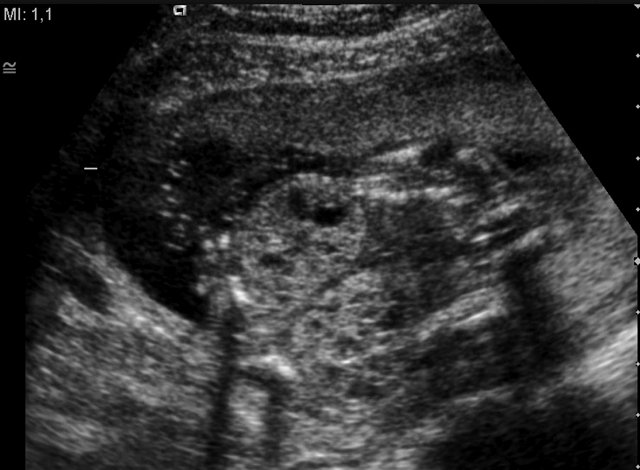
Рис. 3. Увеличенные кистозноизмененные почки плода.
Наличие описанных признаков позволило сформулировать диагноз следующим образом: “Беременность 13,3 недель. Синдром Меккеля-Грубера”.
Пациентке было рекомендовано кариотипирование плода, от которого в этот срок она отказалась. При сроке 16 недель пациентке был выполнен амниоцентез. Кариотип – 46,ХY. Учитывая неблагоприятный перинатальный прогноз при данном синдроме, по желанию пациентки беременность была прервана. Родился плод мужского пола, вес 240 г, длина 21 см. При патологоанатомическом исследовании пренатальный диагноз был подтвержден (рис. 4).
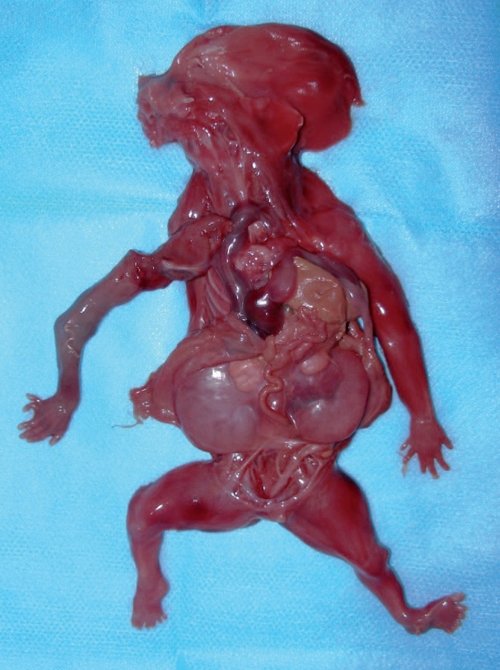
Рис. 4. Фенотип абортуса с синдромом Меккеля-Грубера.
Наблюдение 2
Беременная Е., 26 лет. Данная беременность 4-я, в анамнезе 1 неосложненные роды в срок, 2 самопроизвольных аборта в ранние сроки, 1 беременность, прерванная во II триместре по поводу “патологии почек плода” (результатов патологоанатомического исследования нет). Наследственность супругов не отягощена. Мужу 28 лет, здоров. Брак неродственный, супруги производственных вредностей не имеют.
При эхографии в 14 недель по месту жительства было диагностировано затылочное менингомиелоцеле. В 14-15 недель экспертная эхография проводилась в отделении медицинской генетики МОНИИАГ (Москва) на сканере Accuvix-XQ компании Medison.
При эхографии в срок 14 недель 4 дня фетометрические параметры плода соответствовали гестационной норме. При трансабдоминальном сканировании были обнаружены: затылочное энцефалоцеле размером 10,9х8,8 мм (рис. 5), увеличенные почки повышенной эхогенности (рис. 6). Количество околоплодных вод было нормальным. Был выполнен амниоцентез. Кариотип – 46,ХX.
Рис. 5. Затылочное энцефалоцеле.
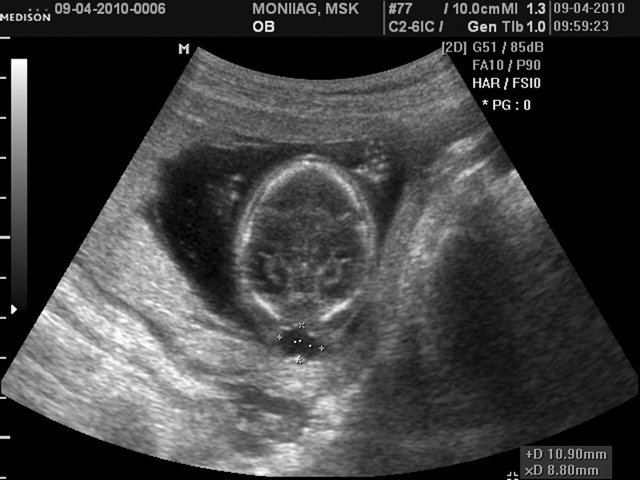
а) Поперечное сечение.
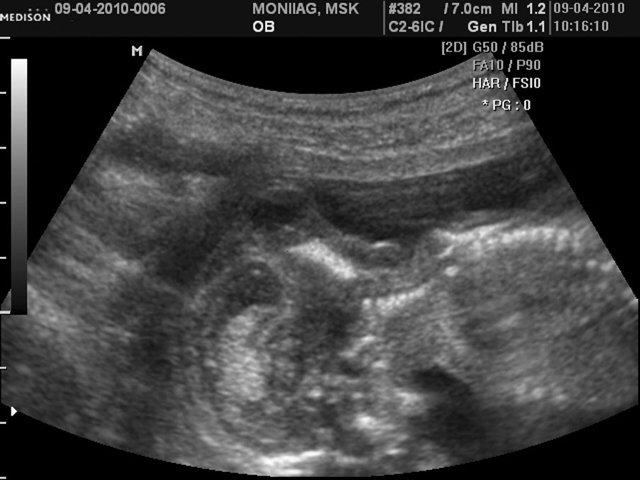
б) Сагиттальное сканирование.
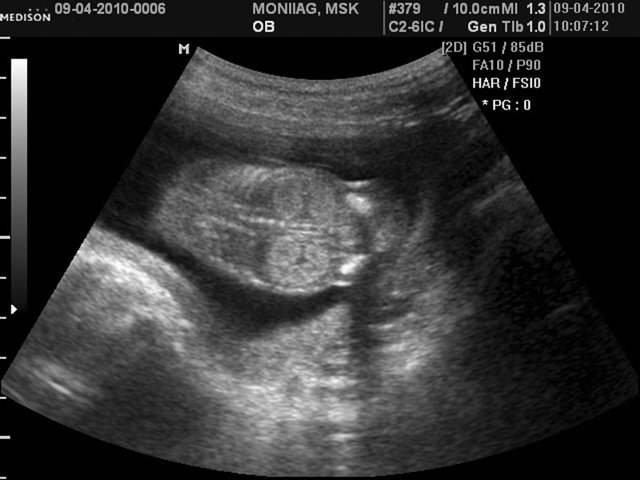
Рис. 6. Увеличенные почки повышенной эхогенности.
Наличие описанных признаков позволило сформулировать диагноз следующим образом: “Беременность 14,4 недель. МВПР плода. Затылочное энцефалоцеле, дисплазия почек. УЗ-маркеры синдрома Меккеля-Грубера”.
Учитывая неблагоприятный перинатальный прогноз при данном синдроме, по желанию пациентки беременность была прервана. Родился плод женского пола, вес 200 г, длина 18 см. При патологоанатомическом исследовании плода пренатальный диагноз был подтвержден (рис. 7).
Рис. 7. Патологоанатомическое исследование плода.
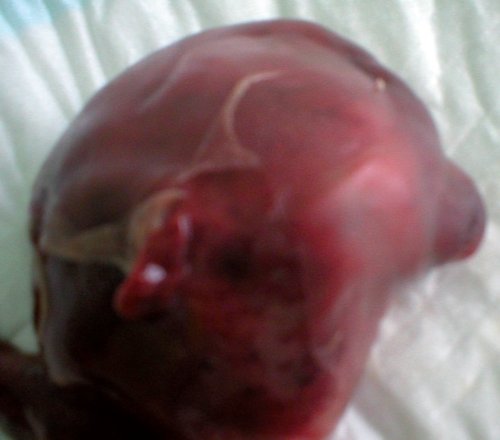
а) Энцефалоцеле у абортуса с синдромом Меккеля-Грубера.
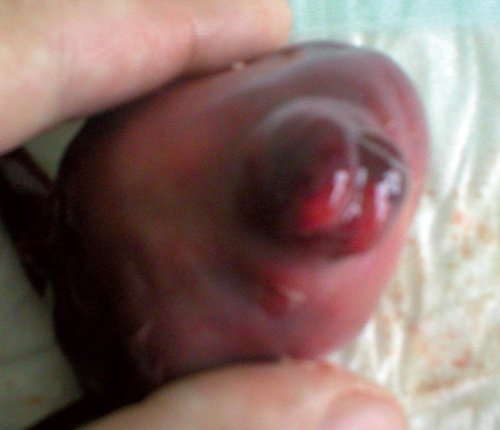
б) Энцефалоцеле у абортуса с синдромом Меккеля-Грубера.
Наблюдение 3
Беременная С., 22 года. Данная беременность первая. Наследственность не отягощена. Мужу 29 лет, здоров. Брак не родственный, супруги производственных вредностей не имеют. При эхографии в I триместре патологии не было выявлено. При эхографии в 22 недели по месту жительства были обнаружены “патологически измененные почки плода”, в связи с чем пациентка была направлена на экспертное исследование в отделение медицинской генетики МОНИИАГ (Москва), эхография проводилась на сканере Accuvix-XQ компании Medison.
При эхографии в 22 недели фетометрические параметры плода соответствовали гестационной норме. Отмечалось выраженное маловодие. При трансабдоминальном сканировании были обнаружены увеличенные в размерах почки плода (рис. 8), в режиме поверхностной объемной реконструкции диагностирована полидактилия обеих стоп (рис. 9).
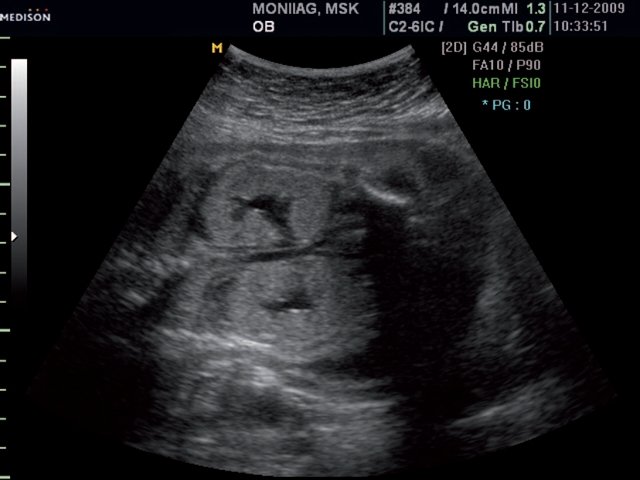
Рис. 8. Увеличенные почки повышенной эхогенности.
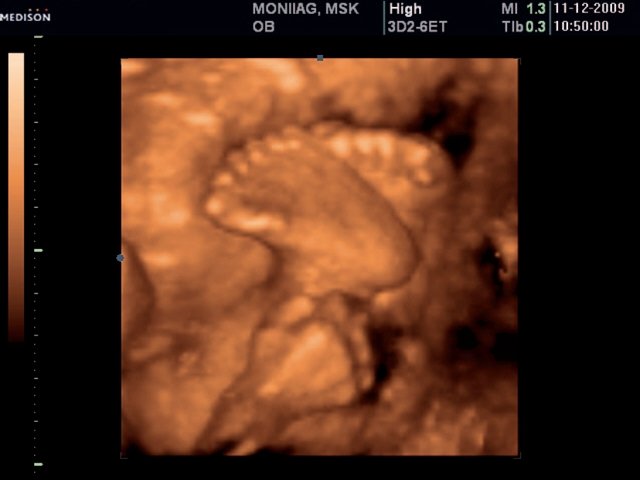
Рис. 9. Стопы плода, полидактилия (режим 3D).
Был выполнен амниоцентез. Кариотип – 46,ХX. Наличие описанных признаков позволило сформулировать диагноз следующим образом: “Беременность 22 недели. МВПР плода. Дисплазия почек, полидактилия обеих стоп. УЗ-маркеры синдрома Меккеля-Грубера”.
Учитывая неблагоприятный перинатальный прогноз при данном синдроме, по желанию пациентки беременность была прервана. Родился плод женского пола, вес 560 г, длина 27 см. При патологоанатомическом исследовании пренатальный диагноз был подтвержден.
Обсуждение
Для синдрома Меккеля-Грубера характерно наличие основных, опорных признаков: затылочной черепно-мозговой грыжи, полидактилии, кистозной дисплазии почек. Мнение Г.И. Лазюка и соавт. [3] о достаточности для постановки ультразвукового диагноза любых двух опорных признаков, с одной стороны, упрощает проблему внутриутробной диагностики этого синдрома. С другой стороны, существует много спорных вопросов в пренатальной идентификации синдрома Меккеля-Грубера.
При обнаружении затылочного менингоцеле, патологически измененных почек и полидактилии необходимо исключить наличие у плода трисомии 13, анеуплоидии с весьма сходными с синдромом Меккеля-Грубера фенотипическими признаками [13].
При скрининговой эхографии в I триместре, во-первых, реальным, а во-вторых, абсолютно обязательным следует считать не только изучение сосудистых сплетений (“бабочки”) головного мозга плода, но и оценку целостности костей черепа. В первых двух наблюдениях, описанных в этой статье, отправной точкой на пути к постановке диагноза было именно выявление энцефалоцеле. Последующая оценка анатомии плода, осмотр почек и кистей/стоп, позволили сформулировать окончательный синдромальный диагноз.
Как показывает опыт, к сожалению, большинство исследователей после обнаружения энцефалоцеле – состояния с исходно неблагоприятным прогнозом, на этом поиск сочетанных аномалий прекращают. Тот же самый подход, к сожалению, практикуется и при выявлении патологии почек плода, когда дискуссия о тактике ведения случая базируется лишь на обсуждении функциональных возможностей почек и не более.
Энцефалоцеле в 14-18% случаев отмечается при хромосомных аномалиях (преимущественно трисомии 13 и 18). Риск нехромосомных синдромов высокий, черепно-мозговые грыжи могут входить в состав многих синдромов: Меккеля-Грубера, амниотических тяжей, фронтоназальной дисплазии, Уокера – Варбурга [16]. Аналогичная ситуация складывается и при дисплазии почек [17]. Большинство из упомянутых синдромов имеют аутосомно-рецессивный тип наследования с риском повторения 25%.
Таким образом, при выявлении в I триместре любой патологии почек показана тщательная оценка кистей и стоп плода с последующим обязательным хромосомным и генным анализом. Аналогичная тактика должна соблюдаться и при наличии в анамнезе данных об энцефалоцеле и дисплазии почек для верификации синдрома Меккеля-Грубера даже в I триместре.
Литература
- OMIM # 249000. Meckel syndrome, typ 1; MKS1. Copyright © 1966-2005. Johns Hopkins University.
- Козлова С.И., Семанова Е., Демикова Н.С., Блинникова О.Е. Наследственные синдромы и медико-генетическое консультирование: Справочник. 2-е изд., дополн. М.: Практика, 1996.
- Лазюк Г.И., Лурье И.В., Черствой Е.Д. Наследственные синдромы множественных врожденных пороков развития. М.: Медицина, 1983.
- Werner H. Meckel syndrome. 2006-02-22-15 // www.theFetus.net
- Auber B., Burfeind P., Herold S. et al. A disease causing deletion of 29 base pairs in intron 15 in the MKS1 gene is highly associated with the campomelic variant of the Meckel – Gruber syndrome // Clin. Genet. 2007. V. 72. N 5. Р. 454-459.
- Лиштван Л.М., Соловьева И.В., Прибушеня О.В. и др. Ультразвуковая диагностика синдрома Меккеля в I триместре беременности // Пренат. Диагн. 2002. Т.1. N 3. С. 193-197.
- Nyberg D.A., Halessy D., Mahony B.S. et al. Meckel – Gruber syndrome: importance of prenatal diagnosis // J Ultrasound Med. 1990. V. 9. P. 691-696.
- Liu S.S., Cheong M.L., She B.Q., Tsai M.S. Firsttrimester ultrasound diagnosis of Meckel – Gruber syndrome // Acta Obstet. Gynecol. Scand. 2006. V. 85. N 6. Р. 757-759.
- Braithwaite J.M., Economides D.L. First-trimester diagnosis of Meckel – Gruber syndrome by transabdominal sonography in a low-risk case // Prenat. Diagn. 1995. V. 15. N 12. Р. 1168-1170.
- Mazneukova V., Kamenov E., Dimitrova L. Ultrasound diagnosis of Meckel-Gruber syndrome at 13 weeks of gestation in families at risk-a case report and literature review // Akush. Ginekol. (Sofiia). 2002. V. 41. N 5. P. 42-45.
- Tanriverdi H.A., Hendrik H.J., Ertan K., Schmidt W. Meckel – Gruber syndrome: a first trimester diagnosis of a recurrent case // Eur. J. Ultrasound. 2002. V. 15. N 1-2. Р. 69-72.
- Sepulveda W., Sebire N.J., Souka A. et al. Diagnosis of the Meckel-Gruber syndrome at eleven to fourteen weeks’ gestation // Am. J. Obstet. Gynecol. 1997. V. 176. N 2. Р. 316-319.
- Weinstein B.J., Benacerraf B.R. Meckel syndrome, first trimester diagnosis. 1994-09-15-12 // www.theFetus.net
- Zurita J. Meckel syndrome, 11 weeks. 2001-06-14-12 // www.theFetus.net
- Manohar S., Karthikeyan M. Meckel Gruber syndrome, two cases. 2009-06-19-08 // www.theFetus.net
- Медведев М.В. Пренатальная эхография. Дифференциальный диагноз и прогноз. 2-е изд. перер. М.: Реал Тайм, 2009.
- Cuillier F., Alessandri J.L., Lemaire P. et al. Renal tubular dysgenesis. 2007-06-25-12 // www.theFetus.net
SonoAce-R7
Универсальный ультразвуковой сканер высокого класса, ультракомпактный дизайн и инновационные возможности.
Источник