
Синдром пагоды фото что это

Агеyезия всего кишечника встречается лишь у аморфных плодов, отсутствие значительной части тонкой кишки – при синдроме «пагоды».
Гипоплазия – укорочение тонкой кншки, при котором протяженность ее менее пятикратной длины тела новорожденного. Структура стенки кишки, диффереицировка на отделы и проходимость не нарушены. Укорочение кишечника, как правило, наблюдается в случае эмбриональных грыж пупочного канатика.
Атрезия и стенозы тощей и подвздошной кишок встречаются с частотой 1 случай на 9828 новорожденных, составляя 52,4% всех случаев атрезии кишечника. Эти пороки обнаруживаются одинаково часто у мужчин и женщин; соотношение атрезии тощей и подвздошной кишок примерно равное, поражаются преимущественно проксимальная часть тощей и дистальиый отдел подвздошной (соответственно в 31 и 36% случаев). Приблизительно в половине случаев наблюдается атрезия в виде свободных слепых концов; менее 40% случаев составляет атрезия с тяжем; мембранозная форма атрезин этой локализации в противоположность двенадцатиперстной кишке отмечается лишь в 13-20% случаев, стенозы встречаются почти в 20 раз реже, чем атрезии. В 6% случаев трезий отмечается множественный их характер. Наиболее частым является сочетание пороков гощей н подвздошной кишок,, крайне редко – тонкой и толстой кишок; комбинация атрезий двеиадцатнперстиой кишки и брыжеечной части кишечника – казуистика.
Атрезия начального отдела тощей кишки (реже терминального отдела двенадцатиперстной) со значительным укорочением остальной части тонкой кишки, которая располагается по спирали в виде винтовой лестницы или гирлянды вокруг оси, образованной брыжеечными сосудами, носит название синдрома «пагоды»или «яблочной кожуры». Частота – 1 случай на 250 000 рождений.
Возникновение атрезий и стенозов брыжеечной части тонкой кишки большинство авторов объясняют нарушением ее кровоснабжения в антенатальном периоде, а также перфорацией ее стенки на ранних стадиях внутриутробного развития. В ряде случаев недоразвитие участка кишки может являться первичным (ТТП – до 6-й недели). Описано возникновение атрезии тонкой кишкн вследствие повреждения ее иглой при амниоцентезе. Морфогенез синдрома «пагоды» связывают с окклюзией верхней брыжеечной артерии на 10-й неделе внутриутробной жизнн. Однако изучение патологической анатомии и кровоснабжения кишечника свидетельствует о том, что в основе данного синдрома лежит агенезия значительной части переднего колена средней кишки эмбриона, а его ТТП не выходит за пределы первых 4 нед развития зародыша.
Врожденные пороки тощей и подвздошной кишки нередко сочетаются с нарушениями поворота кишечника и муковисцидозом. Беременность плодом с такими пороками часто сопровождается многоводием. Атрезии и стенозы тонкой кишки в 39,9 + 3,8% случаев сочетаются с пороками других систем (чаще мочеполовой и опорно-двигательного аппарата); среди комплексов, включающих эти пороки, в 5,4% случаев выявляется трисомия 21 и так же часто – синдромы с аутосомно-рецессивным типом наследования (коротких ребер – полидактилии, Фразера, Меккеля). Атрезия или стеноз тонкой кишки описаны при трисомиях 20 и 21, частичной трисомии lq, Зр, частичной моносомии 20р. Атрезия тощей кишки наблюдалась у 2 снбсов и их теток, что свидетельствует о модифицированном доминантном наследовании. Известны случаи пороков брыжеечной части кишечника у близнецов. Риск для сибсов составляет 0,81 ±0,30%. В отличие от других видов атрезии кишечника синдром «пагоды» так же наследуется по аутосомно-рецессивному типу, как и редкие сочетания атрезии желудка, тонкой и толстой кишки.
Удвоения подвздошной кишки составляют 40%, тощей – 6% от всех удвоений желудочно-кишечного тракта. Здесь могут встречаться все известные разновидности дупликаций. Клинически эти пороки проявляются кишечной непроходимостью и кровотечением. Кистозные формы удвоений следует дифференцировать от кистозиых лимфангиом и кист желточного протока, интралюминальиые дивертикулы – от инвагинатов и ретенционных кист пищеварительных желез стенки кишки. Гетеротопия слизистой оболочки желудка в тощей и подвздошной кишке встречается реже, чем в пищеводе, двенадцатиперстной кишке и меккелевом дивертикуле.
Гетеротопированные фрагменты чаще имеют вид полипозных образований, иногда складок, изредка располагаются экстралюминально. Предполагается, что последний тип может возникнуть при сохранении в стенке кишки остатков омфало-мезентериального протока. Гетеротопия ткани поджелудочной железы чаще обнаруживается в тощей кишке.
Источник
Автор Руслан Хусаинов На чтение 4 мин. Опубликовано 13.12.2020 14:24
Обновлено 13.12.2020 13:12
Синдром Стромма — очень редкое генетическое заболевание, которое проявляется аномалиями развития кишечника, глаз и черепа. Также могут поражаться другие области организма, такие как почки и сердечно-сосудистая система. Генетические мутации в гене CENPF вызывают синдром Стромма. Ген CENPF участвует в регуляции и синтезе ДНК, поэтому мутации в гене влияют на развитие скелета.
Определение
Норвежский педиатр Петтер Стремме (Petter Strømme) впервые описал данный синдром, обследовав двух сестер, родившихся с:
- атрезией тонкой кишки — частичной или полной непроходимостью кишечника
- аномалией глаз, вызывающей проблемы со зрением
- аномалией черепа
В 2007 году клинический генетик Иоланда ван Бевер (Yolande van Bever) предложил название «синдром Стромме» для состояния, поражающего младенцев, рожденных с аналогичными клиническими симптомами.
Причины
Генетические мутации в гене CENPF вызывают синдром Стромма. Данный ген кодирует центромерный белок F, который играет определенную роль в сегрегации хромосом.
Симптомы
Синдром Стромма характеризуется тремя типичными симптомами. Это атрезия кишечника, глазные и черепные аномалии.
Атрезия кишечника
Одним из основных симптомов синдрома Стромма является атрезия тонкой кишки. Младенцы с синдромом Стромма рождаются с атрезией кишечника, которая относится к неполному формированию части тонкой кишки. Атрезия кишечника, также известная как синдром «пагоды» (синдром «яблочной кожуры») — атрезия начального отдела тощей кишки с укорочением остальной части тонкого кишечника, который располагается спирально в виде гирлянды вокруг оси, образованной брыжеечными сосудами. Это может вызвать закупорку кишечника.
Аномалии глаз
Младенцы, рожденные с синдромом Стромма, как правило, имеют недоразвитые глаза или другие аномалии глаз.
- Склерокорнеа — это глазная аномалия, при которой происходит помутнение роговицы, которая сливается с белым наружным слоем глазного яблока (склерой), в результате чего между ними нет четкой границы.
- Микрофтальмия — это состояние, при котором один или оба глаза необычайно малы.
- Микрокорнеа — состояние, при котором одна или обе роговицы необычайно малы.
- Птоз — опущение верхнего века.
- Эпикантус — складки кожи на верхнем веке, которая покрывает внутренний угол глаза.
Аномалии черепа
Другим распространенным симптомом синдрома Стромма является наличие аномалий черепа. Младенцы с синдромом Стромма часто имеют микроцефалию — голова у них намного меньше, чем обычно. Однако не все дети с синдромом Стромма имеют микроцефалию. Некоторые младенцы с этим синдромом имеют обычную окружность головы.
Другие симптомы
Некоторые дети, рожденные с синдромом Стромме, имеют сопутствующие проблемы с почками и сердечно-сосудистой системой.
Диагностика
Врач может диагностировать синдром Стромма, наблюдая за симптомами ребенка. Однако проведение генетического тестирования может дать полное подтверждение наличия синдрома Стромма. Иногда можно диагностировать атрезию кишечника еще до рождения, во время пренатального УЗИ или МРТ плода.
Врач может поставить диагноз после рождения, если присутствуют симптомы атрезии кишечника. Симптомы включают:
- увеличение живота
- отсутствие перистальтики кишечника
- трудности с кормлением
- рвота желчью
Врачи также могут выявлять аномалии черепа и глаз с помощью ультразвукового исследования или МРТ до рождения.
Прогноз
В некоторых случаях синдром Стромма может быть причиной летального исхода в раннем возрасте. Однако у некоторых младенцев общее состояние хорошее. Если врач диагностирует атрезию кишечника до рождения и ребенку быстро проведут лечение, как только он родился, это может снизить риск осложнений.
Лечение
Лечение атрезии кишечника только хирургическое. Ребенку необходимо будет полное парентеральное питание в течение некоторого периода времени после операции. Вводят питательные вещества внутривенно. Это только временно и только до тех пор, пока кишечник ребенка не начнет функционировать правильно.
Заключение
Синдром Стромма — очень редкое генетическое заболевание, которое поражает младенцев, характеризуется тремя основными симптомами: атрезией кишечника, глазными и черепными аномалиями. Однако черепные аномалии присутствуют не у всех детей с синдромом Стромма. Для лечения атрезии кишечника необходимо хирургическое вмешательство. Синдром Стромма может быть смертельным для новорожденных. Однако некоторые дети имеют хорошее общее состояние.
Научная статья по теме: Ученые выявили генетические причины микроцефалии.
Источник
Агеyезия всего кишечника встречается лишь у аморфных плодов, отсутствие значительной части тонкой кишки – при синдроме «пагоды».
Гипоплазия – укорочение тонкой кншки, при котором протяженность ее менее пятикратной длины тела новорожденного. Структура стенки кишки, диффереицировка на отделы и проходимость не нарушены. Укорочение кишечника, как правило, наблюдается в случае эмбриональных грыж пупочного канатика.
Атрезия и стенозы тощей и подвздошной кишок встречаются с частотой 1 случай на 9828 новорожденных, составляя 52,4% всех случаев атрезии кишечника. Эти пороки обнаруживаются одинаково часто у мужчин и женщин; соотношение атрезии тощей и подвздошной кишок примерно равное, поражаются преимущественно проксимальная часть тощей и дистальиый отдел подвздошной (соответственно в 31 и 36% случаев). Приблизительно в половине случаев наблюдается атрезия в виде свободных слепых концов; менее 40% случаев составляет атрезия с тяжем; мембранозная форма атрезин этой локализации в противоположность двенадцатиперстной кишке отмечается лишь в 13-20% случаев, стенозы встречаются почти в 20 раз реже, чем атрезии. В 6% случаев трезий отмечается множественный их характер. Наиболее частым является сочетание пороков гощей н подвздошной кишок,, крайне редко – тонкой и толстой кишок; комбинация атрезий двеиадцатнперстиой кишки и брыжеечной части кишечника – казуистика.
Атрезия начального отдела тощей кишки (реже терминального отдела двенадцатиперстной) со значительным укорочением остальной части тонкой кишки, которая располагается по спирали в виде винтовой лестницы или гирлянды вокруг оси, образованной брыжеечными сосудами, носит название синдрома «пагоды»или «яблочной кожуры». Частота – 1 случай на 250 000 рождений.
Возникновение атрезий и стенозов брыжеечной части тонкой кишки большинство авторов объясняют нарушением ее кровоснабжения в антенатальном периоде, а также перфорацией ее стенки на ранних стадиях внутриутробного развития. В ряде случаев недоразвитие участка кишки может являться первичным (ТТП – до 6-й недели). Описано возникновение атрезии тонкой кишкн вследствие повреждения ее иглой при амниоцентезе. Морфогенез синдрома «пагоды» связывают с окклюзией верхней брыжеечной артерии на 10-й неделе внутриутробной жизнн. Однако изучение патологической анатомии и кровоснабжения кишечника свидетельствует о том, что в основе данного синдрома лежит агенезия значительной части переднего колена средней кишки эмбриона, а его ТТП не выходит за пределы первых 4 нед развития зародыша.
Врожденные пороки тощей и подвздошной кишки нередко сочетаются с нарушениями поворота кишечника и муковисцидозом. Беременность плодом с такими пороками часто сопровождается многоводием. Атрезии и стенозы тонкой кишки в 39,9 + 3,8% случаев сочетаются с пороками других систем (чаще мочеполовой и опорно-двигательного аппарата); среди комплексов, включающих эти пороки, в 5,4% случаев выявляется трисомия 21 и так же часто – синдромы с аутосомно-рецессивным типом наследования (коротких ребер – полидактилии, Фразера, Меккеля). Атрезия или стеноз тонкой кишки описаны при трисомиях 20 и 21, частичной трисомии lq, Зр, частичной моносомии 20р. Атрезия тощей кишки наблюдалась у 2 снбсов и их теток, что свидетельствует о модифицированном доминантном наследовании. Известны случаи пороков брыжеечной части кишечника у близнецов. Риск для сибсов составляет 0,81 ±0,30%. В отличие от других видов атрезии кишечника синдром «пагоды» так же наследуется по аутосомно-рецессивному типу, как и редкие сочетания атрезии желудка, тонкой и толстой кишки.
Удвоения подвздошной кишки составляют 40%, тощей – 6% от всех удвоений желудочно-кишечного тракта. Здесь могут встречаться все известные разновидности дупликаций. Клинически эти пороки проявляются кишечной непроходимостью и кровотечением. Кистозные формы удвоений следует дифференцировать от кистозиых лимфангиом и кист желточного протока, интралюминальиые дивертикулы – от инвагинатов и ретенционных кист пищеварительных желез стенки кишки. Гетеротопия слизистой оболочки желудка в тощей и подвздошной кишке встречается реже, чем в пищеводе, двенадцатиперстной кишке и меккелевом дивертикуле.
Гетеротопированные фрагменты чаще имеют вид полипозных образований, иногда складок, изредка располагаются экстралюминально. Предполагается, что последний тип может возникнуть при сохранении в стенке кишки остатков омфало-мезентериального протока. Гетеротопия ткани поджелудочной железы чаще обнаруживается в тощей кишке.
Источник
Пьебалдизм
Пьебалдизм похож на витилиго, поскольку оба заболевания характеризуются нарушением обмена пигментов и появлением белых прядей волос. Однако пьебалдизм отличается локализацией пятен на коже груди и живота. Кроме того, его признаки — наличие локона белых волос, более темных пятен на лишенной пигмента коже. Эти особенности видны уже с рождения.

Пьебалдизм является редкой наследственной болезнью, а частота ее распространения составляет всего 1 случай на 17 тысяч человек. Эта аномалия никак не влияет на работу внутренних органов и систем.
Анизокория
Анизокория — это состояние, при котором у человека наблюдается разница между размерами зрачков. При этом часто один зрачок имеет патологию, а именно, постоянно находится в зафиксированном положении.

Клинически важно установить, какой именно из зрачков находится в патологическом состоянии, так как это может указывать на наличие других отклонений. Например, отсутствие активности у меньшего из двух зрачков может быть признаком синдрома Горнера, который возникает при поражении нервной системы.
Гетерохромия
Еще одна аномалия, связанная с глазами — гетерохромия. Она выражается в различном цвете радужной оболочки правого и левого глаза или разной окраске определенных участков глаз. Возникает это нарушение от избытка или недостатка меланина — пигмента, который отвечает за цвет. У альбиносов он, к примеру, может вообще отсутствовать, а у людей с гетерохромией наблюдается в измененном состоянии.

Эта аномалия настолько популярна, что многие даже покупают разноцветные линзы, подражая людям с гетерохромией. А еще данная особенность очень привлекает представителей модельных агентств. Мы уже рассказывали о двух братьях из Турции, которые благодаря гетерохромии стали моделями.
Витилиго
О витилиго вы, наверное, уже наслышаны. Сейчас очень много моделей с этим заболеванием, и когда-то мы показывали вам фото самых популярных из них. Витилиго — это нарушение пигментации, которое выражается в исчезновении пигмента меланина на отдельных участках кожи.

Витилиго может начаться в любом возрасте в результате воздействия некоторых лекарственных и химических веществ, после воспалительных и некротических процессов на коже, из-за нервно-трофических, нейроэндокринных и аутоиммунных факторов. А вот предрасположенность к витилиго передается по наследству.
Синдром Шмида-Фраккаро
Синдром Шмида-Фраккаро, который также называют Синдромом кошачьего глаза — редкая врожденная патология, которая возникает из-за наличия дополнительной хромосомы. Один из характерных клинических признаков синдрома — верхняя коломба глаза, то есть отсутствие части радужной оболочки. Этот дефект делает взгляд человека похожим на кошачий, отсюда и название.
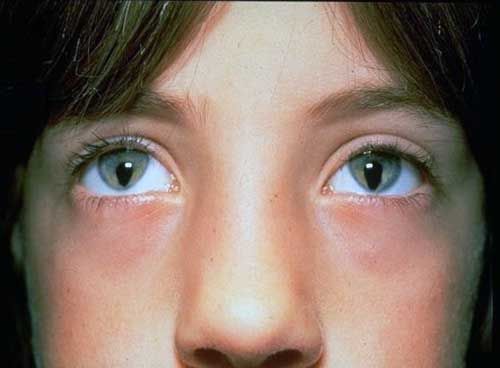
Особенность внешности — далеко не единственное, с чем приходится столкнуться больным синдромом Шмида-Фраккаро. У них могут наблюдаться пороки сердца, низкорослость, аномалии развития почек, умственная отсталость, сколиоз и многие другие проблемы.
Синдром нерасчесываемых волос
Синдром нерасчесываемых волос (еще одно его название — стекловидные волосы) делает его носителей похожими на одуванчики. Их шевелюра торчит во все стороны, при этом возможность уложить ее, собрать в пучок или заплести косу отсутствует в связи с огромным объемом, вызванным структурной аномалией.
Обычно заболевание развивается в грудном возрасте, а в период полового созревания проходит само. Но медицине известны случаи, когда этот синдром преследовал людей на протяжении всей жизни. И хоть сама по себе эта аномалия не представляет никакой опасности, согласитесь, приятного мало.
Смотрите также — «Дети не твои, и ты — не ты», или Как нерожденная женщина стала матерью четырех детей
Понравилось? Хотите быть в курсе обновлений? Подписывайтесь на наш Twitter, страницу в Facebook или канал в Telegram.
Источник
Источник
Синдром Туретта является генетическим заболеванием, которое чаще всего передается от отцов к сыновьям, гораздо реже оно встречается у женщин. При этом женщины, чьи отцы страдали этим недугом, сильно склонны к неврозу навязчивых состояний.
в десятой части случаев синдром Туретта сопровождается еще и копролалией – это болезненная тяга к нецензурной лексике, желание употреблять ругательства тогда, когда это неуместно. Копролалия не является симптомом исключительно данного синдрома, потому что такая проблема встречается и при шизофрении, и при некоторых маниакальных состояниях.
Нельзя сказать, что само заболевание очень редкое – оно встречается от 1 до 10 раз на 2000 людей, чаще всего диагноз ставят в детском возрасте.
Факторы, провоцирующие синдром Туретта
Хотя болезнь и является генетической, некоторые факторы ее активнее провоцируют. Поэтому выделим такие причины синдрома Туретта:
- серьезные инфекции;
- отравление токсинами;
- психические травмы;
- сильные и повторяющиеся стрессы;
- применение психотропных веществ (в том числе неправильное употребление выписанных лекарств).
Как проявляет себя синдром Туретта
К симптомам синдрома Туретта относят:
- разные непроизвольные тики. Подергивания наблюдаются на шее, лице, в области плеч;
- хаотичные и частые движения языка, губ. Причем это может сопровождаться выкрикиванием разных слов, звуков;
- повторение отдельных движений по несколько раз, повторение слогов, слов либо звуков.
По списку видно, что проявления болезни бывают моторными (связанными с движениями) и вокальными (связанными со звуками). Все, что связано с движением, чаще всего проявляет себя с 2 до 8 лет, а вокальные «всплески» характерны и для детей помладше.
Любые тики при таком заболевании не контролируются. Эти признаки синдрома Туретта существуют как бы сами по себе – пациент не в силах с ними совладать. При этом в стрессовой ситуации такие реакции обостряются, становятся более выраженными, яркими и частыми. В условиях полного покоя и расслабления тики беспокоят намного меньше.

Стадии заболевания
Синдром имеет четыре стадии. Определяют их в зависимости от того, как часто происходят тики. Если не чаще одного раз в минуту – речь о первой стадии. Причем пациент часто чувствует, что проблема приближается, и может ее как-то замаскировать. На второй стадии происходит от 2 до 4 тиков в минуту. Это дает сильное напряжение на психику, она перевозбуждается.
Далее процесс идет по нарастающей: на третьей стадии наблюдается 5 и более тиков в минуту, а четвертая характеризуется очень частыми тиками. В последнем случае нередки серьезные психические нарушения, поскольку ЦНС находится в постоянном напряжении.
Как протекает синдром Туретта, есть ли лечение
Полностью вылечить болезнь при помощи конкретных программ нельзя, но нередко она может исчезнуть самостоятельно. Например, такое случается при половом созревании. Если синдром Туретта сохраняется с возрастом, он часто дублирует разные психические расстройства вроде панических атак. Последствий, опасных для жизни, этот синдром в себе не несет, однако ухудшает качество жизни пациента – хотя бы по той причине, что болеющий чувствует себя неловко, не может общаться с окружающими как обычный человек.
Лечение синдрома Туретта предполагает сглаживание выраженных симптомов, повышение качества жизни пациента.
Особенности диагностики
Диагностика синдрома Туретта заключается в комплексе мер:
- опрос пациента, его визуальный осмотр;
- проведение КТ головного мозга;
- проведение МРТ. Выбор конкретного метода исследования делает врач.
Проблемой такого характера занимаются врачи-психиатры, и сам синдром относится к психиатрическим заболеваниям по МКБ-10.
Важно!
Родители не смогут сами определить, какой у ребенка синдром. Дело в том, что есть очень опасные состояния, которые маскируются под тики, поэтому визуально они выглядят так же, как и синдром Туретта. Но убедиться в этом может исключительно профессионал. Если при обследовании специалист заподозрит отклонение, он может назначить ряд дополнительных обследований и анализов (по ситуации). В таком случае нужно обязательно пройти все процедуры и убедиться в точности диагноза.
Чем лечат синдром Туретта в Москве
Все методы, которые используются при лечении подобных заболеваний, можно поделить на медикаментозные и немедикаментозные. Первые предполагают прием седативных препаратов, редко транквилизаторов. Если их и назначают, то короткими курсами и только после того, как врач выяснит, что это необходимо.

Немедикаментозные методы лечения синдрома Туретта:
- расслабляющий массаж – ручной и аппаратный;
- рефлексотерапия. Вариант выбирается в зависимости от ситуации. Такую терапию проводят при помощи рук, специальных игл, электрических импульсов. Смысл в том, чтобы воздействовать на определенные точки тела, которые помогут расслабиться, стабилизировать нервную систему;
- ароматерапия. Она направлена на отдых, успокоение, расслабление. После консультации с врачом ароматерапия доступна и в домашних условиях – это простой и часто действенный метод;
- лечебный сон (электросон). Благодаря импульсам тока через специальные аппараты такой сон нормализует работу мозга, ЦНС. Обычно он длится от 30 минут до 2 часов;
- психотерапия. Она помогает человеку избавиться от тревожности, сложных состояний, психологических травм, а также учит принимать свое состояние.
Важный момент при лечении синдрома Туретта – это правильная обстановка дома, среди близких друзей. Человек с таким отклонением должен получать поддержку, заботу, его необходимо оградить от насмешек и неадекватных реакций на тики.
Народная медицина
При таком недуге можно использовать народные средства, которые направлены на стабилизацию состояния, сохранение спокойствия. Это чаи и настойки из таких трав, как мята, ромашка, мелисса. Можно применять любые успокаивающие сборы, если нет аллергии на компоненты. Однако не стоит рассчитывать на их целебные свойства – это просто легкая поддержка состояния в дополнение к основному лечению (обязательно проконсультируйтесь с лечащим врачом).
Синдром Туретта: вопросы-ответы
Почему синдром так называется?
Он получил свое название в честь Жиля де ла Туретта. Этот врач опубликовал крупный отчет о 9 больных с этим синдромом в 1885 году.
Кто чаще страдает синдромом Туретта?
Симптомы синдрома Туретта чаще встречаются у мужчин – в 3-4 раза чаще, чем у женщин (по разным данным). Также синдром больше присущ детям – половина пациентов к 18 годам от него избавляется (самопроизвольно).
Насколько опасен синдром Туретта?
Прямой опасности жизни и здоровью пациента или окружающих не существует. Все сводится к тому, что из-за неловкости и сложности в общении болеющий начинает замыкаться в себе, избегать людей. Психотерапия и грамотное лечение помогают решить этот вопрос.
При правильном подходе и своевременном лечении синдром Туретта позволяет жить вполне полноценно и без особых проблем. Но для постановки точного диагноза и исключения других, более опасных проблем, обязательно требуется консультация психотерапевта и невролога.
Источник