
Синдром пены шокейра что это

Синдром Пена-Шокейра. Пентада Кантрелла
Синдром Пена-Шокейра (Реnа-Shokeir) является наследственным заболеванием, которое впервые было описано S.D.J. Реnа и М.Н.К. Shokeir в 1974 году. Этот синдром характеризуется артрогрипозом и дисморфическими признаками, возникающими вследствие акинезии плода.
Синонимы. Секвенция акинезии/гипокинезии плода и секвенция фетальных акинетических деформаций.
Этиология. Аутосомно-рецессивная форма наследования заболевания является наиболее распространенной. Имеется несколько описаний необычных проявлений, которые указывают на возможность гетерогенной этиологии.
Риск рецидива. Прогноз риска рецидива бывает неопределен в связи с мультифакториальностью этиологии. В большинстве случаев он находится в пределах от 0 до 25%. Распространенность. Неизвестна.
Патофизиология. Активное шевеление плода начинается с середины первого триместра беременности и имеет основное значение для нормального развития суставов и прилегающих тканей. Отсутствие или уменьшение внутриутробной двигательной активности приводит к тугоподвижности суставов, формированию птеригумов и нарушениям нейромышечных функций со снижением глотательных движений у плода, что вызывает гипоплазию легких и многоводие.
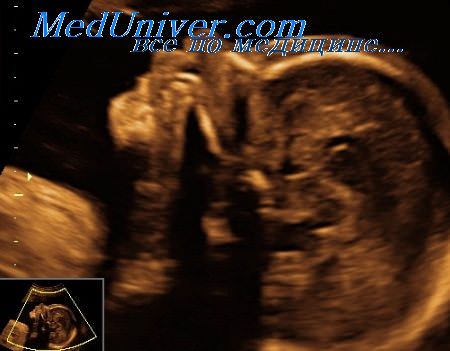
Диагностика. Выявление сочетания патологического положения конечностей, ограниченности движений плода со сниженной или отсутствующей реакцией на акустическую стимуляцию, задержки внутриутробного развития, многоводия и гипоплазии легких позволяет установить диагноз. Также могут выявляться низкое расположение и аномалии ушных раковин, гипертелоризм, короткая шея, расщелина неба, отек мягких тканей головы, деформации грудной клетки, камподактилия и микрогнати. Реже описываемые аномалии при синдроме ПенаШокейра (Pena-Shokeir) включают диафрагмальную грыжу, гастрошизис и микроцефалию.
Дифференциальный диагноз. Трисомия 18 может сопровождаться наличием признаков аналогичных тем, которые обнаруживаются при синдроме Пена-Шокейра (Pena-Shokeir), особенно в отношении черепно-лицевых и внутригрудных аномалий, а также патологии конечностей. Установить диагноз позволяет проведение кариотипирования. Похожее сочетание признаков, за исключением гипоплазии легких, также наблюдается при нелетальных формах артрогрипоза.
Прогноз. Синдром Пена-Шокейра (Pena-Shokeir) является летальным заболеванием. Значительное число пораженных плодов рождается преждевременно. В 30% срочных родов наблюдается мертворождение. Среди выживших большинство детей погибает в течение первых недель жизни. Основной причиной смертности является легочная недостаточность.
Акушерская тактика. Стандартная акушерская тактика ведения беременности изменяется только при возникновении показаний со стороны матери.
Пентада Кантрелла
Данное врожденное заболевание характеризуется двумя основными дефектами: эктопией сердца и дефектом передней брюшной стенки (чаще всего наблюдается омфалоцеле, но может встречаться и гастрошизис) в сочетании с нарушением развития трех, связанных между собою структур: дистального отдела грудины, передней части диафрагмы и диафрагмального отдела перикарда. Имеются сообщения о вариантах классической формы, которые характеризуются неполным проявлением данного синдрома.
Синонимы. Торако-абдоминальная эктопия сердца, эктопия сердца, синдром Кантрелла-Хеллера-Равича (Cantrell-Heller-Ravich); синдром пентады и перитонеоперикардиальная диафрагмальная грыжа.
Распространенность. Встречается очень редко. В литературе имеются сообщения приблизительно о 90 клинических наблюдениях и еще меньшем их числе с полным подтверждением имевшегося синдрома.
Этиология. Неизвестна. Иногда может сочетаться с хромосомными аномалиями.
Риск рецидива. Неизвестен. Данных о рецидивах не имеется. Опубликовано сообщение об одном случае конкордантного развития синдрома у монозиготных близнецов.
Диагностика. Наличие полной формы дизрупции порока развития передних стенок грудной клетки и брюшной полости, которая сопровождается пятью признаками, описанными J.R. Cantrell et al., является классической формой пентады и может быть выявлена при ультразвуковом исследовании начиная с середины второго триместра. Обнаружение сокращающегося вне пределов грудной клетки сердца в сочетании с омфалоцеле позволяет установить диагноз пентады. Для исключения хромосомных аномалий рекомендуется проведение кариотипирования. Вследствие компрессии органов как грудной клетки, так и брюшной полости могут возникать асцит и гидроторакс.
Генетические нарушения. Неизвестны.
Патогенез. Остановка развития сегмента латеральной мезенхимы в период от 14-го до 18-го дня после зачатия приводит к незакрытию брюшной стенки и неполному слиянию наружных первичных тяжей.
Сочетанные аномалии. Является правилом наличие внутрисердечных аномалий (таких, как тетрада Фалло (Fallot), дефекты межжелудочковой перегородки). Другие нарушения включают в себя черепно-лицевые аномалии, гидроцефалию, анэнцефалию, незавершенный поворот толстой кишки, косолапость и хромосомные аберрации.
Дифференциальный диагноз. Изолированная торакальная эктопия сердца, эктопия сердца в сочетании с синдромом амниотических перетяжек, аномалия развития стебля тела, изолированное омфалоцеле, синдром Беквита-Видемана (Beckwith-Wiedeman) и хромосомные аномалии.
Прогноз. Выживание является исключительным случаем и зависит от размера дефекта абдоминальной стенки, степени тяжести поражения сердца и наличия сочетанных аномалий. В редких случаях при наличии легких форм возможно проведение хирургической коррекции пороков. В тех ситуациях, когда имеется полная экструзия сердца и органов брюшной полости, прогноз исключительно неблагоприятный.
Акушерская тактика. Родоразрешение рекомендуется проводить в специализированных перинатальных медицинских центрах.
– Также рекомендуем “Синдром Пфайффера. Диагностика и прогноз синдрома Пфайффера”
Оглавление темы “Врожденные синдромы плода”:
1. Синдром Клиппеля-Треноне-Вебера. Синдром Ларсена у плода
2. Синдром летального птеригума. Лиссэнцефалия I типа
3. Синдром Мекеля. Диагностика и прогноз при синдроме Мекеля
4. Мезомелическая дисплазия. Диагностика и прогноз мезомелической дисплазии
5. Микроцефалический первичный нанизм. Синдром Неу-Лаксова
6. Синдром Нунан у плода. Секвенция маловодия
7. Несовершенный остеогенез. Диагностика и прогноз несовершенного остеогенеза у плода
8. Синдром Пена-Шокейра. Пентада Кантрелла
9. Синдром Пфайффера. Диагностика и прогноз синдрома Пфайффера
10. Синдром Поланда. Секвенция Поттер и синдром prune-belly
Источник
Несовершенный остеогенез. Диагностика и прогноз несовершенного остеогенеза у плода
Данная гетерогенная группа генетических заболевний (разделенная на четыре типа, определенных Силленсом (Sillence): I, II, или врожденный, III и IV) характеризуется выраженной хрупкостью костей, ведущей к патологической оссификации и множественным переломам. Имеются следующие признаки при каждом их четырех типов заболевания.
• Тип I не характеризуется пренатальными изменениями, и диагноз устанавливается после рождения, когда начинают появляться деформации конечностей.
• Тип II является наиболее тяжелой формой. Она проявляется множественными нарушениями развития скелета, такими как укорочение и угловые искривления костей и появление выступов на их поверхностях вследствие множественных переломов, а также деминерализацией костей свода черепа, узкой и колоколообразной грудной клеткой в результате переломов ребер и уменьшением двигательной активности плода.
• Тип III является менее тяжелым, чем тип II, и обычно проявляется множественными переломами при рождении с развитием прогрессирующих деформаций костей от неонатального периода до подросткового возраста. Эта форма заболевания диагностируется начиная со второго триместра беременности при типе NIC.
• Тип IV характеризуется наименее выраженными проявлениями нарушений и пренатально не определяется. Заболевание обычно сопровождается преждевременным развитием остеопороза в возрасте 40-50 лет.
Синонимы. Врожденный несовершенный остеогенез, синдром Ван дер Хове (Van der Hoeve), болезнь Лобстейна (Lobstein), меловые хрупкие кости, болезнь хрупких костей, болезнь Вролика (Vrolik).
Распространенность. Частота возникновения составляет 0,4 на 10 000 новорожденных и примерно половина из них (0,19 на 10 000) имеет тип II.
Этиология. В большинстве случаев возникновение типов I и IV обусловлено аутосомно-доминантным наследованием. Тип II формируется в результате новой доминантной мутации (с несколькими сообщениями об аутосомно-рецессивном наследовании), и тип III образуется аутосомно-рецессивно или аутосомно-доминантно.
Риск рецидива. Зависит от формы наследования заболевания. В целом риск рецидива составляет от 2 до 5% для типа II в тех случаях, когда это не вызвано спонтанной мутацией.
Диагностика. Эхографические признаки, которые могут выявляться, особенно при типе II, включают увеличение ширины, укорочение и переломы длинных трубчатых костей, которые имеют выступы на поверхности, образованные костными мозолями; снижение оссификации костей свода черепа, улучшение визуализации внутричерепных структур, уменьшенную в размерах и колоколообразную грудную клетку, аномальную форму мозгового черепа; широкие ребра неравномерной толщины, искривление длинных трубчатых костей и патологию лицевых структур. Типичным признаком несовершенного остеогенеза II типа является возможность визуализировать оба кортикальных слоя костей. Обычно дистальный по отношению к датчику слой попадает в зону «акустической тени», формирующейся позади проксимального слоя.
Однако при несовершенном остеогенезе этот феномен отсутствует вследствие выраженной деминерализации (аналогичные признаки могут выявляться при ахондрогенезе и гипофосфатазии). Большинство аномалий может быть выявлено пренатально с помощью эхографии начиная с первой половины второго триместра беременности. Нормальные эхографические данные у пациенток из группы риска по развитию заболевания у плода полностью не исключают наличие диагноза. В большинстве случаев если в пренатальном периоде переломы не возникают, то диагноз обычно устанавливается только после рождения.
Генетические нарушения. Причиной заболевания являются мутации одного из двух генов, ответственных за синтез коллагена I типа (COL1А1 и COL1A2).
Патогенез. Дефекты синтеза, организации и химического строения коллагена I типа, который присутствуете коже, связках, сухожилиях деминерализованных костях и дентине, являются причиной снижения минерализации и возникновения хрупкости костей.
Сочетанные аномалии. Кифосколиоз, глухота, мышечная гипотония, паховые грыжи, гидроцефалия, водянка и задержка внутриутробного развития.
Дифференциальный диагноз. Гипофосфатазия (инфантильная форма), ахондрогенез и другие заболевания, сопровождающиеся нанизмом и укорочением конечностей.
Прогноз. Несовершенный остеогенез II типа всегда летален, и наиболее частыми причинами гибели являются, вероятно, внутричерепные кровоизлияния и дыхательная недостаточность. Другие формы развиваются после рождения и сопровождаются прогрессирующими деформациями длинных трубчатых костей и наличием низкого роста.
Акушерская тактика. Вследствие летальности заболевания при диагностике несовершенного остеогенеза II типа прерывание беременности может быть предложено на любом сроке гестации. Для других форм прерывание предлагается до наступления периода жизнеспособности плода. Если отдается предпочтение пролонгированию беременности, стандартная пренатальная тактика ведения не изменяется.
– Также рекомендуем “Синдром Пена-Шокейра. Пентада Кантрелла”
Оглавление темы “Врожденные синдромы плода”:
1. Синдром Клиппеля-Треноне-Вебера. Синдром Ларсена у плода
2. Синдром летального птеригума. Лиссэнцефалия I типа
3. Синдром Мекеля. Диагностика и прогноз при синдроме Мекеля
4. Мезомелическая дисплазия. Диагностика и прогноз мезомелической дисплазии
5. Микроцефалический первичный нанизм. Синдром Неу-Лаксова
6. Синдром Нунан у плода. Секвенция маловодия
7. Несовершенный остеогенез. Диагностика и прогноз несовершенного остеогенеза у плода
8. Синдром Пена-Шокейра. Пентада Кантрелла
9. Синдром Пфайффера. Диагностика и прогноз синдрома Пфайффера
10. Синдром Поланда. Секвенция Поттер и синдром prune-belly
Источник
Множественный врожденный артрогрипоз (МВА) у ребенка: причины, клиника, диагностика, лечениеТермин множественный врожденный артрогрипоз (MBA) часто ошибочно применяют к гетерогенной группе расстройств, отличительная черта которых — врожденные контрактуры нескольких суставов. Иногда используется широкое определение «множественные врожденные контрактуры». Некоторые авторы, однако, отмечают, что термин MBA должен применяться только к очень специфическому, точно определенному состоянию, описанному ниже (Mennen et al, 2005). Выявлено большое количество причин или их комбинаций, вызывающих множественные врожденные контрактуры (Hall, 1997), но часто этиология остается неизвестной. Движения плода необходимы для его нормального развития, и общий фактор, вызывающий множественные врожденные контрактуры — это снижение или отсутствие подвижности плода. Термин синдром деформации вследствие фетальной акинезии (FADS) в настоящее время применяется к нескольким фенотипам, являющимся результатом сниженной внутриутробной подвижности. FADS диагностируется при УЗИ даже в первом триместре беременности (Witters et al., 2002). Общие признаки FADS, не считая контрактур, включают краниофациальные аномалии, микрогнатию, птеригии конечностей, гипоплазию легких и задержку внутриутробного развития. FADS может развиваться в результате любой причины, вызывающей снижение подвижности плода, включая экзогенные факторы (например, присутствие материнских антител к ацетилхолиновым рецепторам), ограничение пространства плода (например при маловодий), и патологию центральной нервной системы или периферической нервно-мышечной системы. Клинические проявления и течение болезни крайне вариабельны и зависят от продолжительности периода снижения подвижности, количества и локализации обездвиженных суставов, и наличия или отсутствия аномалий головного мозга и другой патологии. К менее тяжелым формам относятся некоторые синдромы дистального артрогрипоза. а) Врожденная амиоплазия – это наиболее часто встречающаяся форма FADS с характерной клинической картиной, для которой применим узкий термин MBA и которую иногда называют нейропатической формой врожденной амиоплазии (Flail et al., 1983). Заболевание спорадическое, частота встречаемости от 1 на 3000 до 1 на 10000 живых новорожденных (Hall et al., 1983; Mennen et al., 2005), на эту патологию приходится около трети всех случаев множественных врожденных контрактур (Hall, 1997). При патологоанатомическом исследовании в переднем роге выявляется уменьшение количества больших двигательных нейронов, но с минимальным глиозом, более мелкие нейроны переднего рога присутствуют или их число увеличено (Hageman et al., 1987b), в отличие от спинальной мышечной атрофии, при которой отмечается снижение количества нейронов обоих типов. Мышечная ткань замещается фиброзно-жировой. При ЭМГ могут наблюдаться признаки денервации, хотя часто эти признаки не выражены или ниже порога чувствительности, что указывает на стабильное состояние, при котором мышцы или пучки уже фиброзировались и стали электрически «немыми», либо остались нормальными. По данным литературы обычно наблюдается сниженная подвижность и тазовое предлежание плода, а родоразрешение нередко выполняется с помощью кесарева сечения. При родах часто случаются переломы (Sells et al., 1996). В большинстве, но не во всех случаях симметрично поражаются все конечности, снижается мышечная масса, характерны покатые плечи, руки ротированы внутрь, разогнуты в локтях, согнуты в лучезапястных суставах, пальцы также согнуты, тазобедренные и коленные суставы в положении сгибания, обычно отмечается тяжелая эквиноварусная деформация стоп. Иногда имеется подвывих или вывих бедра. Может сохраняться некоторая пассивная подвижность, и в таких случаях выявляется паралич мышц, в норме осуществляющих движения в пораженном суставе. Сгибательные борозды обычно отсутствуют или аномальны, что указывает на раннее начало болезни во внутриутробном периоде, часто имеются аномальные углубления. Плоская капиллярная гемангиома — в большинстве случаев по средней линии лица, но иногда в других областях — отмечается в 80-90% случаев (Sells et al., 1996). Часто наблюдается микрогнатия, примерно у четверти пациентов имеются контрактуры височно-нижнечелюстного сустава. Выраженные трудности при кормлении в младенческом возрасте отмечались у 51 из 87 пациентов, по данным Robinson (1990). Впоследствии обычно развиваются нарушения речи и задержка роста. Интеллект обычно нормальный. Для достижения оптимальных результатов лечения большое значение имеют раннее физиотерапевтическое лечение под наблюдением специалиста, раннее хирургическое лечение, реабилитация и тщательное наблюдение (Mennen et al., 2005). Несмотря на обширные деформации, исход относительно хороший. Из 38 детей, обследованных Sells et al. (1996), две трети в возрасте около пяти лет были полностью способны к самообслуживанию, и многие — полностью или относительно самостоятельны. Большинство из них перенесли от пяти до семи ортопедических манипуляций, шинирование.
б) Дистальный артрогрипоз. Дистальный артрогрипоз поражает преимущественно кисти и стопы. Были предложены различные классификации (Hall et al., 1982а), но последняя редакция предполагает, 10 форм заболевания. Во всех случаях нужно сопоставлять время публикации сообщений с изменяющейся номенклатурой. Категории включают пальце-таранный дизморфизм, синдромы Фримана-Шелдона, Гордона, тризм-псевдокамптодактилии, врожденную контрактурную арахнодактилию и доминантный птеригиум-синдром (Beals, 2005). Обычно наблюдаются деформации верхних конечностей: ульнарная девиация, камптодактилия, гипоплазия или отсутствие сгибательных борозд и «накладывающиеся» пальцы; тогда как часто встречающиеся аномалии нижних конечностей — эквиноварусная деформация стопы, пяточно-вальгусная деформация, варусное приведение стопы (metatarsus varus) и вертикальное положение таранной кости (Beals, 2005). Симптомы артрогрипоза являются общими признаками, тогда как другие проявления болезни, например аномалии лица, отличаются у разных больных. У некоторых пациентов удается выявить генетическую причину (Beals, 2005). Часто выявляется доминантное наследование с различной пенетрантностью, но это не единственный механизм наследования. Большое количество хромосомных и генетических комплексных синдромов, проявляющихся множественными врожденными контрактурами, дисморфизмом и мультисистемными аномалиями также сопровождаются FADS (Hall, 1997). Наиболее частый из них — фенотип Пена-Шокейра (Репа и Shokeir, 1974; Hageman et al., 1987a), включающий в себя гипоплазию легких, лицевой дизморфизм и различные аномалии головного мозга. Фенотипу Пена-Шокейра может сопутствовать патология как периферической нервно-мышечной, так и центральной нервной систем (Kho et al., 2002). Другие интересные варианты фенотипа Пена-Шокеейра — FADS включают в себя финский летальный синдром врожденных контрактур (LCCS тип 1) и другую разновидность — израильских кочевников (Israeli Bedouin variety) (LCCS тип 2). Финский вариант — аутосомно-рецессивное заболевание приводит к летальному исходу до наступления 32 недели гестации и локализовано в зоне хромосомы 9q34 (Vuopala et al., 1994; Makela-Bengs et al., 1998). LCCS2 (хромосома 12q13 зона) приводит к летальному исходу в неонатальном периоде, отличительным признаком является выраженное растяжение мочевого пузыря (Narkis et al., 2004). Легкий аутосомно-рецессивный нейрогенный тип в большой израильско-арабской инбредной (вырождающейся, вследствие родственных браков) семье был картирован в зоне 5qter (Tanamy et al., 2001); также описана Х-сцепленная спинальная мышечная атрофия младенцев с артрогрипозом (Хр 11,3-q11.2) (Kobayashi et al., 1995). Другие формы нейрогенного артрогрипоза — это локализованные формы, как, например, при spina bifida или агенезии крестца, и редкий тип, при котором поражается только шея и верхние конечности, вероятно, вследствие сегментарной патологии спинного мозга (Darwish et al., 1981; Hageman G. et al., 1993). в) Другие артрогрипозы. Часто встречаются другие фенотипы с аномалиями ЦНС, как изолированные, так и сопутствующие заболеваниям клеток переднего рога (Fedrizzi et al., 1993). Заболевание может вызываться как деструктивными процессами (Perlman et al., 1995), так и мальформациями (Hageman et al., 1985, 1987b, 1988b; Massa et al., 1988; Baker et al., 1996). Может развиваться внутричерепная кальцификация (Ilium et al., 1988). Также в числе причин могут быть другие редкие прогрессирующие синдромы (Hageman et al., 1988а, b; Di Rocco et al., 1995). Множественный врожденный артрогрипоз (врожденная амиоплазия, АМС), вызванный патологией клеток переднего рога (Fedrizzi et al., 1993; Hall, 1997) отличается от спинальной мышечной атрофии (SMA) 1 типа (болезнь Верднига-Гофмана), при которой могут развиваться контрактуры, но обычно нетяжелые. Тем не менее, существует вариант SMA 1 типа с тяжелым артрогрипозом и врожденными переломами. У шести из 12 пациентов с нейрогенными проявлениями АМС (Burglen et al., 1996) и еще у одного больного с патологией спинного мозга, совпадающей с SMA (Garcla-Cabezas et al., 2004) была выявлена делеция гена выживаемости двигательного нейрона 5q13 (SMN), что позволяет предположить существование подгруппы АМС, аллельной SMA. Однако в отличие от обычного клинического течения нейрогенной АМС в постнатальном периоде, у пациентов с делецией SMN болезнь прогрессирует и приводит к летальному исходу на первом месяце жизни, что больше напоминает фенотип SMA. 1. Артрогрипоз, вызванный мышечными заболеваниями. Мышечные заболевания, в основном врожденные дистрофии, но также и врожденные миопатии, являются диагностируемыми причинами множественных врожденных контрактур (Banker, 1994). В длинном перечне миопатических процессов присутствуют также и метаболические расстройства (Swoboda et al., 1997). Множественные контрактуры часто наблюдаются при врожденной форме миотонической дистрофии (Shore, 1975), являясь одним из неблагоприятных прогностических признаков (Connoly et al., 1992). Случай артрогрипоза с рваными красными мышечными волокнами и недостаточностью комплекса I (Laubscher et al., 1997) показывает, что в некоторых случаях нужно искать митохондриальную цитопатию. 2. Заболевания периферических нервов. Поражения периферических нервов редко являются причиной множественных врожденных контрактур (Yuill и Lynch, 1974; Charnas et al., 1988; Boylan et al., 1992). При транзиторной неонатальной форме миастении может наблюдаться FADS с врожденными контрактурами, особенно в случае выработки материнских антител к ацетилхолиновым рецепторам плода. Хотя эта патология встречается редко, правильная диагностика имеет большое значение не только из-за риска развития рецидива, но также потому, что это заболевание потенциально поддается терапии (Polizzi et al., 2000). Легкие врожденные контрактуры могут наблюдаться при рано манифестирующей врожденной миастении (Burke et al., 2004) и при некоторых мутациях субъединиц ацетилхолиновых рецепторов (Brownlow et al., 2001). Так как множественные контрактуры являются скорее симптомом, а не заболеванием, определение механизма наследования зависит от точной диагностики специфического синдрома (Hall, 1997). Возможны все формы наследования. Это могут быть доминантные наследственные формы артрогрипоза, аутосомно-рецессивные синдромы и Х-сцепленные синдромы, в том числе, и картированный на Xp11.3-q12, быстро приводящий к летальному исходу (Hall et al., 1982b; Kobayashi et al., 1995). – Также рекомендуем “Аномалии мышц у ребенка: агенезия, гипоплазия, гипертрофия, кривошея, травма” Редактор: Искандер Милевски. Дата публикации: 16.1.2019 Оглавление темы “Мышечные расстройства у детей.”:
|

Источник