
Синдром при котором недоразвита рука

Синдром Холта-Орама, также называемый синдромом «сердце-рука», является наследственным заболеванием, которое характеризуется нарушениями верхних конечностей и сердца. Холт и Орам впервые описали это состояние в 1960 году при обследовании пациентов с дефектами межпредсердной перегородки и аномалиями большого пальца.
Синдром Холта-Орама. Причины
Синдром Холта-Орама является генетическим расстройством, которое имеет аутосомно-доминантный признак.
- Дефектный ген находится на длинном плече хромосомы 12.
- Молекулярно-генетические исследования показывают, что болезнь вызывается мутациями, которые инактивируют фактор транскрипции TBX5.
- Спорадическая болезнь может развиваться De Novo при мутации в гене TBX5.
Синдром Холта-Орама. Патофизиология
Синдром наследуется по аутосомно-доминантному типу. Заболевание связано с мутациями в гене транскрипционного фактораTBX5, что в конечном итоге выливается в развитие аномалий сердца и верхних конечностей. Патофизиологические последствия являются прямым результатом развития пороков развития сердца и верхних конечностей. Информация по экологическим факторам, которые могут нести ответственность за развитие синдрома, не поступала.
Синдром Холта-Орама. Симптомы и проявления
Верхние конечности.
Хотя клинические проявления показывают переменный характер, аномалии верхних конечностей у детей присутствуют всегда. Аномалии могут быть односторонними или двусторонними, асимметричными и симметричными. Аплазия, гипоплазия, или другие аномальные проявления в этих костях проявляют целый спектр фенотипов (от простых аномалий и до полного отсутствия пальцев). Иногда порок развития конечностей может быть настолько серьезным, что у ребенка может развиться фокомелия (порок развития, при котором будут отсутствовать проксимальные участки рук). Наиболее распространенным проявлением синдрома являются пороки развития или слитые кости запястья. Кистевые аномалии являются единственными типами проявлений этого синдрома, которые присутствуют у каждого пациента с синдромом. Однако эти аномалии, у некоторых людей, могут быть обнаружены только при прохождении рентгенологических процедур.
Сердце.
Примерно 75% больных имеют некоторую сердечную аномалию. У большинства пациентов присутствует либо дефект межпредсердной перегородки либо дефект межжелудочковой перегородки, эти дефекты могут варьироваться в размере и расположении. Сердечные аномалии также могут включать в себя сердечные дефекты проводимости и фибрилляции предсердий. Эти аномалии часто присутствуют даже в отсутствие дефектов перегородки.
Синдром Холта-Орама. Фото

Полидактилия всех четырех конечностей.

Обратите внимание на мизинцы.

Синдром Холта-Орама у матери.

И у её сына.

Гипоплазия конечностей.

Слева рентген грудной клетки до операции. Правый желудочек, правое предсердие и центральная часть легочной артерии расширенна. Справа, рентген грудной клетки после оперативного закрытия дефекта межпредсердной перегородки, у пациента наблюдается значительное сокращение правого предсердия, желудочка и сокращение растяжения легочной артерии.

Проявления синдрома Холта-Орама.
Пупочная грыжа с полидактилией левой верхней конечности.

Рентген грудной клетки показывает кардиомегалию.

На снимке виден дополнительный палец.
Физическое обследование
- Деформации верхних конечностей
- Всегда присутствуют, но могут быть односторонними или двусторонними
- Левосторонние аномалии часто более тяжелые, чем аномалии правой руки
- Неравные длины рук происходят в связи с аплазией, гипоплазией, слиянием или аномальным развитием радиальной кости, запястья
- Аномалии предплечья
- Могут отсутствовать пальцы
- Аномалии большого пальца
- Ограничения в движениях плечевым суставом
- Фокомелия
- Поражения сердца
- Брадикардия
- Нерегулярный пульс (эктопия)
- Мерцательная аритмия
- Легочный систолический шум
- Голосистолический шум
Синдром Холта-Орама. Диагностика
- Рентгенография костей рук
- Рентгенография грудной клетки. Результаты могут продемонстрировать увеличенные легочные артерии в связи с развитой легочной гипертензией или кардиомегалией, также по результатам можно найти свидетельства застойной сердечной недостаточности.
- Эхокардиография. На этой процедуре можно определить присутствие дефектов перегородки или другие сердечные аномалии. Наиболее распространенной сердечной аномалией является дефект межжелудочковой перегородки. Некоторые пациенты также могут иметь ВСД. Необъяснимое, значительное расширение правого предсердия у плода может означать наличие синдрома Холта-Орама.
- ЭКГ для определения участия проводящей системы.
- Генетическая оценка также имеет важное значение — мутации в гене TBX5 обнаруживаются у около 75% лиц, отвечающим строгим клиническим критериям синдрома Холта-Орама.
Синдром Холта-Орама. Лечение
Медикаментозное лечение пациента может выполняться, как правило, как в амбулаторных, так и в стационарных условиях, хирургическое вмешательство также может быть необходимым. Пациентам с блокадой сердца может потребовать установка постоянного кардиостимулятора. Хирургическое вмешательство может быть выполнено для коррекции пороков сердца или, возможно, для улучшения функций конечностей.
Хирургическое вмешательство.
Большинство пороков сердца, такие как дефект межпредсердной перегородки либо дефект межжелудочковой перегородки поддаются хирургической коррекции, если у пациента не наблюдается легочная гипертензия или коллапс желудочка. Сегодня доступно несколько хирургических методов, которыми можно исправлять сердечные аномалии, к ним также можно отнести имплантацию чрескожных транскатетерных устройств, которыми можно закрыть отверстие в перегородке.
- Септальные дефекты без гемодинамически значимых аномалий не требуют коррекции.
- Дети с тяжелыми аномалиями конечностей могут быть направлены к ортопедами для рассмотрения и проведения специальных коррекционных процедур.
- Дети с укороченными конечностями могут извлечь выгоду из протезов.
Синдром Холта-Орама. Осложнения
- Сердечная недостаточность
- Аритмия
- Блокада сердца
- Мерцательная аритмия
- Инфекционный эндокардит
- Внезапная смерть
Синдром Холта-Орама. Прогноз
Прогноз, как правило, хороший, но он сильно зависит от тяжести сердечных пороков.
Источник
Синдром Холта-Орама – это очень редкая и серьезная генетическая патология, передающаяся по наследству от поколения к поколению и имеющая аутосомный тип наследования. Такое заболевание является очень необычным, так как способно поражать сразу несколько жизненно важных генов. Эти гены влияют на правильное формирование скелета, а также на функциональные особенности сердечной мышцы. В этой статье мы рассмотрим, что же представляет собой синдром Холта-Орама, а также узнаем о методах его диагностики, причинах развития и основных методах лечения. Данная информация поможет вам разобраться во всех особенностях протекания данного недуга.
Синдром Холта-Орама: что собой представляет
Данное заболевание предупредить невозможно, так как оно имеет наследственный характер. Патология опасна не только тем, что нарушает работу сердца, но также и тем, что оставляет свой отпечаток на состоянии верхних конечностей. При этом чем больше страдает сердце, тем хуже будет развиваться и скелет. Именно поэтому является очень важным распознать синдром Холта-Орама как можно раньше, для того чтобы была возможность предотвратить самые тяжелые последствия. С этой целью проводится просто огромное количество различных генетических исследований, на фоне которых и можно определить точный диагноз. Также проведения различных методов диагностики позволяет определить, будет ли возникать патология и у будущих поколений.

Каждая беременная женщина во время вынашивания плода в обязательном порядке должна проходить ультразвуковое обследование. Такая процедура поможет уже на ранних стадиях определить, будет ли иметь малыш отклонения в строении его костной ткани. Если же таковые имеются, врачи должны проверить и состояние сердца будущего малыша. Однако чаще всего определить симптомы болезни можно только после того, как ребенок родится.
Какими бывают врожденные дефекты
Синдром Холта-Орама, фото которого вы сможете посмотреть на данном ресурсе, чаще всего накладывает свой отпечаток на строение именно верхней части скелета. Обычно пациенты страдают различными аномалиями кистей или предплечий, которые могут поражать как одну сторону тела, так и две одновременно. При этом такие аномалии могут иметь самый разнообразный характер. Например, на месте одного пальца у пациента может быть сразу две или три фаланги. Бывают случаи, когда большие пальцы на руках отсутствуют полностью.
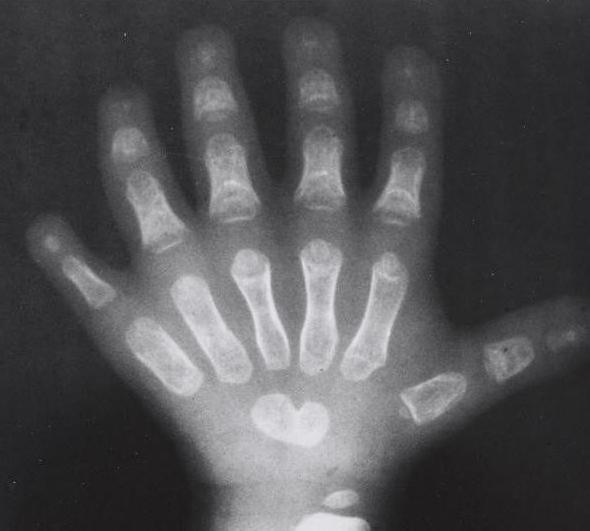
Чаще всего такое явление наблюдается именно на правой руке. Существует большое количество аномалий, касающихся такой патологии. Медицине известны случаи, когда малыши рождались без предплечий вовсе, или же их лучевые кости были недоразвитыми. Также болезнь может давать о себе знать искривлением позвоночника или неправильно сформированной грудиной, или ключицами. Чаще всего недоразвитые конечности доставляют пациентам только эстетические неприятности. Однако стоит учитывать, что заболевание также отражается и на здоровье сердечной мышцы. Практически все пациенты, страдающие синдромом Холта-Орама (синдром рука-сердце), имеют также патологии в развитии сердечной мышцы. При этом заболевания могут носить как легкий характер, так и достаточно тяжелый. Не исключены случаи развития сердечной недостаточности.
Как наследуется данная патология
Синдром Холта-Орама, описание которого вы можете прочитать в этой статье, носит доминантно-аутосомный тип наследования. Это говорит о том, что патология будет регулярно передаваться от поколения к поколению. Однако в некоторых случаях поврежденный ген не обладает высокой проявляемостью, а это говорит о том, что болезнь будет проявляться не у каждого поколения, а через одно. Чаще всего недуг передается непосредственно от родителей к детям.
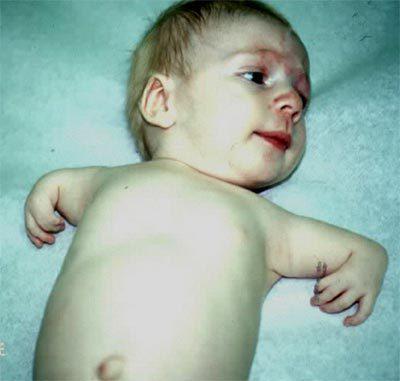
Но бывают случаи, когда ген начинает мутировать даже при условии, что у малыша родители были абсолютно здоровыми. Даже в этом случае болезнь будет передаваться следующим поколениям. Заболевание может передаваться независимо от того, кто оказался носителем мутированного гена, мужчина или женщина.
Синдром Холта-Орама: причины развития патологии
Как было сказано выше, чаще всего болезнь передается по наследству, от родителей к малышу. Обычно данное заболевание поражает двенадцатую хромосому. Однако недуг может возникнуть и при возникновении мутации данного гена даже у здоровых родителей.
Основная симптоматика
Согласно исследованиям специалистов, клинически заболевание может носить переменный характер. Но вот патологии верхних конечностей у малышей будут присутствовать в любом случае. При этом аномалии могут возникать как в одной части человеческого тела, так и сразу в двух. Они могут быть асимметричными или же симметричными. Аномалии в строении скелета будут присутствовать у каждого пациента, страдающего синдромом Холта-Орама (лечение заболевания будет описано ниже). У некоторых пациентов такие аномалии можно заметить только при проведении рентгенологического исследования.

Также большое количество пациентов будут иметь и заболевания сердца. Чаще всего пациенты страдают недугами, связанными с межпредсердной перегородкой или с межжелудочковой перегородкой.
Дефекты сердечной мышцы
Если при возникновении этого недуга страдает межпредсердная перегородка, то очень часто это может приводить к возникновению порока сердца, а также к другим заболеваниям, которые возникают внутри столь жизненно важного органа. Такая патология возникает в том случае, когда канал связи между левым и правым предсердием остается полностью открытым. Порок сердца врожденного характера приводит к возникновению сильной отдышки, повышенной утомляемости, а также к снижению защитных сил организма. Синдром Холта-Орама примерно в пятнадцати случаев сопровождается пороком сердца.
Проведение диагностики
Диагностические критерии синдрома Холта-Орама позволяют точно определить разновидность патологии. Для того чтобы точно определить масштабы заболевания, каждый пациент должен сделать рентген костей рук, а также грудной клетки. С помощью такого метода обследования можно увидеть патологии легких, а также развитие сердечной недостаточности.
С помощью эхокардиографии специалисты смогут установить наличие различных аномалий сердечной мышцы. Также очень важно сделать электрокардиограмму для того, чтобы определить состояние проводящей системы.
Ну и, конечно же, очень важно пройти генетическое обследование, с помощью которого можно обнаружить мутацию определенных генов.
Основные методы лечения
Обычно консервативное лечение с помощью применения различных медикаментозных препаратов может проводиться как в домашних условиях, так и в стационаре. Также очень часто проводится хирургическое вмешательство, целью которого является улучшить внешний вид конечностей и подправить здоровье сердечно-сосудистой системы. На сегодняшний день большинство заболеваний сердца, которые возникают при наличии такого недуга, как синдром Холта-Орама, можно устранить при помощи проведения оперативного вмешательства.
Могут ли возникать осложнения
На самом деле очень часто данный недуг приводит к большому количеству осложнений со стороны сердечно-сосудистой системы и опорно-двигательного аппарата. Нередко на фоне синдрома Холта-Орама пациенты страдают сердечной недостаточностью, аритмией, блокадой сердца и другими заболеваниями, которые могут привести и к летальному исходу. Обычно врачи дают хорошие прогнозы на выздоровление, однако они будут зависеть, в первую очередь, от того, насколько тяжелыми являются сердечные пороки.
Диагностика врожденного порока сердца
Если у новорожденного малыша существуют подозрения на развитие столь серьезного заболевания, как порок сердца, то очень важно своевременно вызвать кардиолога, для того чтобы максимально точно поставить диагноз и выбрать методы лечения данного заболевания.

В этом случае будет проведен целый комплекс диагностических мер, позволяющих определить само заболевание, а также причины его развития. Синдром Холта-Орама, патогенез которого описан в нашем обзоре, довольно часто сопровождается врожденным пороком сердца. При этом во внутриутробном периоде такую патологию удается заметить далеко не всегда.
Основные симптомы порока сердца
Чаще всего понять, что малыш родится с пороком сердца, несложно. При наличии такой патологии цвет кожи ребенка, а также его губы и уши имеют синеватый оттенок. Иногда ребенок, наоборот, становится слишком бледным, а его нижние конечности очень холодные. Если болезнь имеет врожденный характер, то обычно ее можно определить уже сразу после появления малыша на свет. Однако иногда болезнь вовсе не дает о себе знать до начала полового развития, после чего здоровье ребенка начнет ухудшаться, и все процессы развития будут протекать гораздо медленнее.
Выводы
Очень важно внимательно ознакомиться с такой патологией, как синдром Холта-Орама. Фильмы документального плана о людях, страдающих данным заболеванием, позволят понять, что это не приговор. Многие родители, думают, что их малыш, страдающий генной мутацией, не будет жить счастливой полноценной жизнью. Не стоит забывать о том, что медицина не стоит на месте. С помощью проведения различных типов хирургического вмешательства можно эстетически улучшить состояние конечностей малыша, а также увеличить их функциональность. Для этого используются специальные протезы и другие приспособления.

Также не стоит забывать, что синдром Холта-Орама в большинстве случаев сопровождается наличием внутренних заболеваний сердца. Поэтому при наличии дефектов скелета еще во внутриутробном периоде очень важно проверить также сердце малыша. Некоторые заболевание незначительные, поэтому не требуют кардинальных методов лечения. Другие же являются достаточно опасными, поэтому нуждаются в проведении хирургического вмешательства. В любом случае врачи дают очень хорошие шансы на то, что пациенты, страдающие синдромом Холта-Орама, смогут иметь достаточно хороший уровень жизни. Для этого очень важно вовремя выявить патологию и начать своевременно ее лечить.
Начните заботиться о своем здоровье, а также о здоровье ваших детей прямо сейчас. И тогда вам не будут страшны никакие заболевания. Будьте здоровы, любите себя и всегда верьте в то, что жизнь прекрасна. И она принесет вам много счастья и радости.
Источник
Синдром Холта – Орама – это тяжелое заболевание с аутосомным типом наследования, которое характеризуется поражением сразу нескольких генов. Они отвечают за анатомическое строение сердца и скелета. Этот недуг называют синдромом «рука-сердце».
Что это такое?
Это заболевание, передаваемое по наследству, сочетает в себе варьирующие пороки развития верхних конечностей и врожденные пороки сердца. Очень важно распознать болезнь в начале ее развития. Для этого осуществляют проведение генетических исследований. Они не только подтверждают диагноз, но и прогнозируют вероятность возникновения заболевания в будущих поколениях.

Ультразвуковое исследование в период беременности дает возможность своевременно обнаружить нарушения в развитии скелета. При соответствии показаний признакам заболевания проводится внутриплодное сканирование сердца будущего ребенка. Обычно синдром Холта – Орама определяется после рождения малыша. Подозрительные дефекты в строении скелета тщательно обследуются кардиологом, так как тяжесть заболевания зависит от сердечных проявлений синдрома.
Врожденные дефекты заболевания
Характерны аномалии в строении предплечья и кисти, которые проявляются с одной стороны или с обеих сразу. Степень недоразвития и деформации конечностей бывает разной: это наличие трех фаланг на одном пальце или когда полностью отсутствуют большие пальцы на руках. Чаще всего это бывает с пальцами на левой руке. Встречаются тяжелые случаи, когда наблюдается гипоплазия лучевой кости или полное ее отсутствие. Гораздо реже отмечают: сросшиеся между собой пальцы, деформацию грудины, недоразвитие ключиц и лопаток, искривление позвоночника. Скелетные изменения не представляют угрозы для жизни человека. Но нарушение функциональной способности пораженного болезнью органа осложняет жизнь.
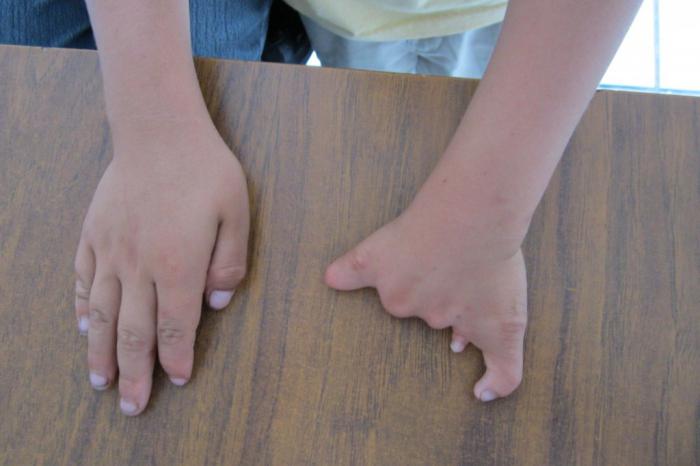
Около 85% пациентов страдают врожденными пороками сердца: сужением и открытым протоком аорты, стенозом легочной артерии, нарушением строения клапана. Часто встречается врожденный дефект межпредсердной перегородки. Он бывает разным по тяжести: от еле различимого до обширного вторичного дефекта, последствием которого является сердечная недостаточность.
Аутосомно-доминантный тип наследования
Для этого заболевания характерны:
- Регулярная передача недуга из одного поколения в другое.
- Если ген отличается высокой проявляемостью, заболевание не перескакивает через поколение, а прослеживается у членов семьи в каждом роду.
- Ген передается от родителей детям. Исключение составляют впервые появившиеся случаи заболевания в семье из-за спонтанной мутации гена, который впоследствии передается также от больного предка к здоровому потомку.
- Чаще заболевание передается по одной родительской линии.
- Не все в роду наследуют ген заболевания: у здоровых членов семьи рождаются потомки без признаков проявления недуга.
- Если у детей одна мать, а отцы разные, или наоборот, отец один, а матери разные, дети общего родителя могут унаследовать болезнь.
- Аутосомно-доминантный тип наследования не зависит от принадлежности человека к определенному полу. Мужчины и женщины поражаются недугом одинаково.
Межпредсердная перегородка
Если наблюдается ее дефект, развивается врожденный порок сердца, который может быть изолированным или сочетаться с другими заболеваниями внутри сердца. Дефект межпредсердной перегородки развивается тогда, когда образуется открытое сообщение между двумя предсердиями: левым и правым. Если у больного заболевание синдром Холта – Орама, у него появляется одышка, шумы в сердце, повышается утомляемость и отставание в физическом развитии, наблюдается бледность кожи и частые респираторные заболевания. Врожденный порок сердца, обусловленный такой патологией, наблюдается примерно у 5-15% больных, причем у женщин встречается в два раза чаще, чем у мужчин.
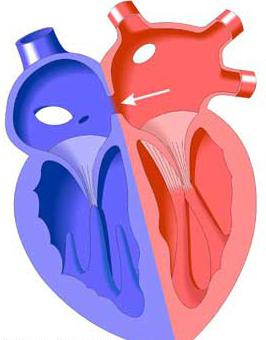
Дело в том, что плод, находясь в утробе матери, имеет в межпредсердной перегородке открытое отверстие овальной формы. Оно обеспечивает нормальное кровообращение. После того как ребенок родится, отверстие у большинства новорожденных закрывается. Хотя бывают случаи, когда этого не происходит. Но сброс крови через это отверстие такой незначительный, что человек не чувствует аномалии и не подозревает даже, что она у него есть. Он живет спокойно до самой старости. Такие наследственные аномалии, как дефект межпредсердной перегородки, могут быть различных размеров, формы и встречаются в разных ее отделах.
Межпредсердные дефекты. Типы
- Аномалия первичной части перегородки является разновидностью неправильного развития подушечек и локализуется в нижней части ее трети рядом с трикулярными клапанами. Они, в свою очередь, могут деформироваться и не справляться с выполнением предназначенной функции. Бывают случаи, когда с их помощью образуется общий с предсердно-желудочковой перегородкой клапан.
- У межпредсердной перегородки бывает дефект ее вторичной части, который наблюдается вверху или в области ямки овальной. В этом случае кровь в правое предсердие поступает из левого. Часто эти дефекты называют вторичными и возникают они при наличии недостатка ткани перегородки. Но при этом сохраняется функциональная и анатомическая проходимость дефекта.
Симптомы межпредсердных дефектов
- Синдром Холта – Орама напрямую связан с возрастом больного, размером дефекта перегородки, величиной сопротивления легочных сосудов. Многие пациенты с данным пороком выглядят абсолютно здоровыми и не предъявляют жалоб. Дело в том, что кровь может сбрасываться слева направо из-за чрезмерной физической нагрузки или утомляемости, а не из-за дефекта. В подавляющем большинстве случаев шум в сердце не прослушивается. Это иногда случается и при значительном дефекте межпредсердной перегородки.
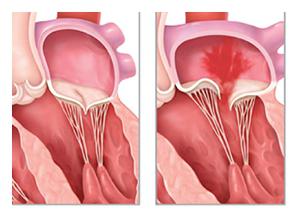
- Эта аномалия часто становится причиной пролапса митрального клапана. У пациентов с таким заболеванием хорошо прослушивается шум или клик.
- Если размеры дефекта очень большие, у человека появляется тахикардия, одышка, сердечный горб, когда границы органа расширены в правую сторону. Можно легко, при помощи пальцев, определить пульсацию артерии легочной.
- Вторичный дефект межпредсердной перегородки в редких случаях может быть причиной сердечной недостаточности. Это наблюдается у 3-5% пациентов, преимущественно у детей до одного года. Если не сделать операцию, риск летального исхода велик.
Межпредсердные дефекты. Причины
Аномалии происходят тогда, когда недоразвиты первичная или вторичная перегородки и зндокардиальные валики в период эмбрионального развития.
- Возникновение дефекта связано с генетическими, физическими, экологическими и инфекционными факторами.
- Аномальное развитие перегородки ребенка в утробе матери имеет большую степень риска в семьях, чьи родственники больны врожденным пороком сердца.
- Заболевания вирусного характера беременной женщины могут стать причиной дефекта межпредсердной перегородки. Это краснуха, герпес, ветряная оспа, сифилис.
- Лекарственные препараты и алкоголь во время беременности.
- Вредные вещества на производстве, радиационное облучение.
- Осложнения из-за токсикозов, в результате которых вынашивание ребенка ставится под угрозу.
Дефекты межпредсердной перегородки. Классификация
В период от третьей до восьмой недель беременности в утробе матери закладывается сердце плода. Если образуется неполная перегородка между правой и левой половинками сердца, это приводит к тому, что возникают дефекты межпредсердной перегородки, которые различают по размерам, количеству и месту расположения отверстий. Классификация врожденных пороков сердца такова:
- Первичные дефекты характеризуются значительными размерами: от трех до пяти сантиметров. Местом его локализации является нижняя часть перегородки, которая располагается выше предсердно-желудочковых клапанов, как раз над ними. У них нет нижнего края.
- Вторичные дефекты образуются в результате недоразвития перегородки вторичной. Они небольших размеров и находятся в центральной ее части или в районе устьев вен полых. Бывает, что дефекты перегородки и аномальное впадение в правое предсердие легочных вен сочетаются. В этом случае межпредсердная перегородка сохраняется в нижней части.
- Наблюдаются случаи, когда вторичный и первичный дефекты присутствуют одновременно.
- Межпредсердная перегородка может вообще отсутствовать. Это приводит к образованию общего, единственного предсердия, которое называют трех камерным сердцем.
Врожденный порок сердца. Диагностика
Если у новорожденного подозревается это заболевание, необходим срочный вызов кардиолога. Специалист рассматривает симптомы заболевания, оценивает характер пульса и давления, состояние всех органов и систем. После осмотра младенца проводится электрокардиограмма, ультразвуковое исследование, фонокардиограмма, рентген сердца. В случае тяжелой формы заболевания диагностика врожденных пороков дополняется еще одним обследованием – катетеризацией сердца, когда в его полость вводят зонд.

Очень часто родители малыша возмущаются, что лечащий врач не выявил это опасное заболевание, когда будущая мать весь срок беременности наблюдалась у него. Причиной врожденного порока сердца может быть:
- Низкий уровень профессионализма врача.
- Врожденные заболевания не всегда удается вовремя диагностировать из-за особенности строения сердца и сосудов плода.
- Несовершенство медицинского оборудования.
Причины врожденных пороков сердца
Врожденные пороки сердца у новорожденных являются следствием анатомических дефектов сердца, его клапанов и сосудов, которые могут возникать еще в утробе матери. Поначалу они могут быть незаметными, а могут беспокоить ребенка сразу после его рождения. Пороки сердца – это самые распространенные врожденные пороки у новорожденных и составляют около 30%. Они являются наиболее частой причиной смерти. Причины развития дефекта межпредсердной перегородки:

- Перенесенные во время беременности вирусные заболевания, такие как краснуха, корь и другие.
- Генетическая мутация, вызванная употреблением будущей матерью наркотиков, алкоголя, никотина.
- Различные отклонения хромосомных наборов родителей.
- Облучение ионизирующей радиацией.
- Дефицит микроэлементов и витаминов в рационе питания беременной женщины.
- Воздействия лекарственных препаратов на плод.
Симптомы врожденного порока сердца
Рожденные с таким заболеванием малыши отличаются. У них кожные покровы, губы, ушные раковины имеют синеватый или голубой цвет. Иногда наоборот, кожа малыша побледнеет, а ножки становятся холодными. Это называется белым пороком новорожденных. Бывают случаи, когда при выслушивании специалист отчетливо слышит шумы в сердце.
Очень часто врожденное заболевание органа незаметно сразу же после появления малыша на свет. В течение 10 лет ребенок будет выглядеть совершенно здоровым. Но, потом болезнь начнет проявляться в виде побледнения или посинения кожи. Станут заметными отклонения в физическом развитии, появится одышка.
Источник