
Синдром рокитанского или синдром тестикулярной феминизации

")
Синдром тестикулярной феминизации (Синдром Морриса) – наследственное заболевание, характеризующееся развитием женских половых признаков при наличии мужского кариотипа (XY). Симптомы этой патологии обладают широким спектром выраженности – от фенотипически полноценной женщины до полноценного мужчины с целым рядом промежуточных вариантов, на чем построена классификация данного состояния. Диагностика синдрома тестикулярной феминизации производится на основании результатов гинекологического или урологического осмотра, ультразвуковых исследований органов малого таза, изучения кариотипа и молекулярно-генетических анализов. Специфического лечения этого заболевания не существует, для улучшения качества жизни больных применяются разнообразные хирургические вмешательства.
Общие сведения
Синдром тестикулярной феминизации – генетическое заболевание, поражающее лиц с мужским кариотипом и приводящее к развитию у них разнообразных по выраженности женских половых признаков вплоть до полной феминизации. Нарушения подобного типа регистрировались давно, сразу после открытий хромосомных основ пола и исследований кариотипа, но данное заболевание впервые описал американских гинеколог Джон Моррис в 1953 году. Он изучил известных на тот момент (более 80) пациентов с мужским псевдогермафродитизмом и выявил ряд семейных случаев, что позволило ему определить синдром тестикулярной феминизации как X-сцепленное рецессивное наследственное заболевание. В некоторых источниках эту патологию можно найти под названием синдрома Морриса. В настоящий момент установлено, что встречаемость синдрома тестикулярной феминизации составляет примерно 1 случай на 20-60 тысяч новорожденных мужского пола, однако частота носительства патологического гена среди женщин неизвестна. Данное заболевание является причиной почти 20% случаев мужского псевдогермафродитизма и обуславливает заметную долю от всех разновидностей первичной аменореи.
Синдром тестикулярной феминизации (Морриса)
Причины синдрома тестикулярной феминизации
Исследования в области современной генетики позволили выявить молекулярно-генетические механизмы синдрома тестикулярной феминизации – таковыми оказались мутации гена AR, локализованного на X-хромосоме. Продуктом экспрессии данного гена является белок-рецептор к тестостерону и его метаболитам (в основном, дигидротестостерону), наличие которого и обеспечивает реакцию организма на мужские половые гормоны. На сегодняшний день выявлено более 300 различных типов мутаций гена AR, приводящих к синдрому тестикулярной феминизации. Все они имеют рецессивный характер, поэтому женщины (по кариотипу), имеющие гомологичную Х-хромосому, могут выступать только в качестве носителя и передавать это заболевание своим сыновьям с вероятностью 50%.
Из-за нарушений в структуре гена AR кодируемый им белок-рецептор получается дефектным, в зависимости от типа мутаций его реакция на воздействие тестостерона и сходных с ним соединений изменяется. При наиболее тяжелом течении синдрома тестикулярной феминизации рецептор становится совсем неспособным взаимодействовать с мужскими половыми гормонами, поэтому клетки организма теряют к ним чувствительность, сохраняя ее к эстрогенам (в основном, к эстрадиолу). Это приводит к развитию организма полностью по женскому типу при наличии и функционировании яичек. Некоторые другие типы синдрома тестикулярной феминизации обусловлены сохранением чувствительности к тестостерону, но на крайне низком уровне, что становится причиной широкого спектра клинических проявлений. Активность клеток Сертоли, выделяющих тестостерон, при любой форме синдрома тестикулярной феминизации сохраняется и может быть даже несколько повышена.
Крайняя форма синдрома тестикулярной феминизации имеет следующий патогенез – еще на этапе эмбрионального развития из-за отсутствия влияния тестостерона по причине нечувствительности к нему организма происходит формирование женских половых органов. Образуется «слепое» влагалище, расщепленная мошонка становится большими половыми губами, зачатки фаллоса – клитором. На этапе полового созревания также полностью отсутствует реакция клеток на тестостерон, поэтому при синдроме тестикулярной феминизации (полный тип) ткани организма испытывают влияние только женских половых гормонов. Данное обстоятельство приводит к формированию выраженных вторичных половых признаков – пышной груди, «модельных» форм, распределению жировой клетчатки по женскому типу, тонкому голосу. Однако менструации и, тем более, возможность беременности в этом случае полностью исключены, так как у больных отсутствует матка и необходимые для менструального цикла гормональные факторы.
Классификация и симптомы синдрома тестикулярной феминизации
Синдром тестикулярной феминизации характеризуется обширным спектром выраженности проявлений и разновидностей заболевания, для их систематизации была разработана специальная классификация данной патологии. В первую очередь, все формы этого состояния делятся на две группы – полные и неполные. Полная форма синдрома тестикулярной феминизации возникает при полном отсутствии чувствительности организма к тестостерону и характеризуется полноценным женским фенотипом. Обычно рождается здоровая девочка, не имеющая, на первый взгляд, каких-либо отклонений в развитии. С началом полового созревания такие больные нередко становятся весьма красивыми (с «модельной внешностью») из-за превалирующего влияния эстрогенов. Однако при этом у них отсутствует оволосение в области подмышечных впадин и на лобке, не наступает менархе – зачастую именно аменорея в возрасте 14-16 лет становится поводом для обращения к врачу, где в рамках гинекологического исследования и выявляется синдром тестикулярной феминизации.
Неполная форма синдрома тестикулярной феминизации характеризуется намного более разнообразной клинической картиной. Как правило, причиной этой формы патологии выступают дефекты рецепторов к тестостерону, не приводящие к полной потере чувствительности, а только к ее значительному снижению или нарушению. Согласно классификации, принятой в 1996 году, выделяют пять основных форм или степеней неполного синдрома тестикулярной феминизации:
1. 1-я степень (мужской тип) характеризуется типично мужским фенотипом без каких-либо отклонений. В крайне редких случаях может наблюдаться высокий голос и признаки гинекомастии в подростковом возрасте. При этом практически всегда нарушены процессы сперматогенеза, поэтому у больных данным типом синдрома тестикулярной феминизации наблюдается мужское бесплодие.
2. 2-я степень (преимущественно мужской тип) – данный вариант заболевания проявляется более выраженными нарушениями вирилизации и формирования половых органов, хотя фенотипически больные являются мужчинами. У них часто обнаруживается гипоспадия, возможно развитие микропениса или сочетание этих признаков. Пациенты с этим вариантом синдрома тестикулярной феминизации нередко имеют гинекомастию и неравномерное отложение подкожной жировой клетчатки.
3. 3-я степень (амбивалентный тип) – характеризуется выраженным уменьшением полового члена, который становится похожим на клитор. Мошонка разделена настолько, что ее половины напоминают большие половые губы, часто наблюдается гипоспадия, крипторхизм. При этом типе синдрома тестикулярной феминизации также отмечается расширение таза, гинекомастия, относительно узкие плечи.
4. 4-я степень (преимущественно женский тип) – при этой форме синдрома тестикулярной феминизации больные являются женщинами по своим фенотипическим признакам, однако их клитор часто гипертрофирован, урогенитальный синус формирует короткое слепое влагалище. Нередко при этом варианте заболевания наблюдается такое нарушение, как сращение половых губ.
5. 5-я степень (женский тип) – больные этой формой синдрома тестикулярной феминизации фенотипически являются женщинами, практически никаких признаков вирилизации у них не обнаруживается за исключением несколько увеличенного размера клитора. В период полового созревания он может увеличиваться еще больше, достигая размеров микропениса.
У больных синдромом тестикулярной феминизации также часто возникают паховые грыжи из-за нарушения проходимости яичек по паховому каналу. Осложнениями гипоспадии могут быть разнообразные воспалительные процессы в мочевыделительной системе (уретриты, пиелонефриты). Крипторхизм грозит в будущем злокачественным перерождением тканей яичка, что является наиболее тяжелым осложнением данного заболевания.
Диагностика и лечение синдрома тестикулярной феминизации
Основными методами диагностики этого состояния являются гинекологический или урологический осмотр, ультразвуковое исследование, изучение наследственного анамнеза, молекулярно-генетический анализ и определение уровня половых гормонов. Раньше всего удается диагностировать неполные формы синдрома тестикулярной феминизации 2-5 степеней, так как нарушения в строении половых органов заметны уже при рождении ребенка. При осмотре врач-неонатолог может заподозрить наличие генетического заболевания и назначить дополнительные уточняющие исследования. Сочетание таких пороков развития с нормальным или даже повышенным уровнем тестостерона в крови и крипторхизмом говорит в пользу наличия синдрома тестикулярной феминизации.
Неполная форма заболевания 1 степени и полный тип патологии в большинстве случаев определяются намного позднее. Поводом для обращения мужчин (по своим фенотипическим признакам) с синдромом тестикулярной феминизации к специалистам часто служит подозрение на бесплодие. Анализ спермы при этом выявляет азооспермию, в анамнезе больного присутствует крипторхизм (нередко устраненный оперативным путем), паховые грыжи. Подтвердить наличие синдрома тестикулярной феминизации в этом случае возможно только при помощи генетической диагностики. Полная форма заболевания чаще всего диагностируется в 14-15 лет, когда девушки обращаются к специалисту из-за отсутствия менструаций. При гинекологическом осмотре у них определяется «слепое», закрытое в верхней трети влагалище, ультразвуковые исследования обнаруживают отсутствие матки и ее придатков. При этом могут выявляться семенники, находящие на различных этапах спуска в мошонку – располагающиеся в пределах брюшной полости, пахового канала, изредка в половых губах.
Концентрация тестостерона при синдроме тестикулярной феминизации соответствует уровню здорового мужчины или даже несколько превышает его. При этом количество эстрогенов не достигает нижней отметки нормы для девушек аналогичного возраста. Изучение наследственного анамнеза может обнаружить признаки Х-сцепленной передачи заболевания. Молекулярно-генетическая диагностика синдрома тестикулярной феминизации производится врачом-генетиком при помощи автоматического секвенирования последовательности гена AR или других методик. Возможно также выявление носительства патологической формы гена у здоровых женщин и пренатальная диагностика этого заболевания.
Лечение синдрома тестикулярной феминизации полного типа часто ограничивается простым удалением семенников с последующей коррекцией гормонального фона (при необходимости). Это требуется для профилактики семиномы и других форм злокачественного перерождения тканей яичка. Такие больные, с детства воспитывающиеся как девочки, после постановки диагноза могут нуждаться в психологической помощи. Терапия синдрома тестикулярной феминизации неполного типа характеризуется большим количеством пластических операций для воссоздания привычного вида половых органов, груди, оптимального функционирования мочевыделительной системы. Больные с любой формой этого заболевания бесплодны, что также зачастую требует помощи психологов.
Прогноз и профилактика синдрома тестикулярной феминизации
Прогноз синдрома тестикулярной феминизации относительно выживаемости больных довольно благоприятный – при полной форме патологии больные могут прожить нормальную жизнь женщины, обладая при этом мужским кариотипом. Неполные формы при правильно проведенной хирургической коррекции нарушений и пороков развития зачастую также не приводят к тяжелым и жизнеугрожающим осложнениям. Особенно высокую угрозу при синдроме тестикулярной феминизации представляет рак яичек в случае крипторхизма, поэтому его необходимо устранять – выводом семенников в мошонку (при мужском фенотипе) или удалением желез (в случае женского фенотипа). Профилактика синдрома тестикулярной феминизации производится посредством генетического выявления носительства патологического гена при отягощенном наследственном анамнезе и в случае подтверждения – пренатальной диагностикой этой патологии.
Источник
Причины синдрома
Специалисты рассматривают две основные причины развития синдрома: наследственную и эмбриопатическую.
Наследственная теория подтверждается случаями семейного заболевания синдромом Рокитанского-Кюстнера. Чаще всего семейная форма сопровождается множественными аномалиями развития (почек, позвоночника, сердца). При этом заболевание передается по аутосомно-доминантному механизму.
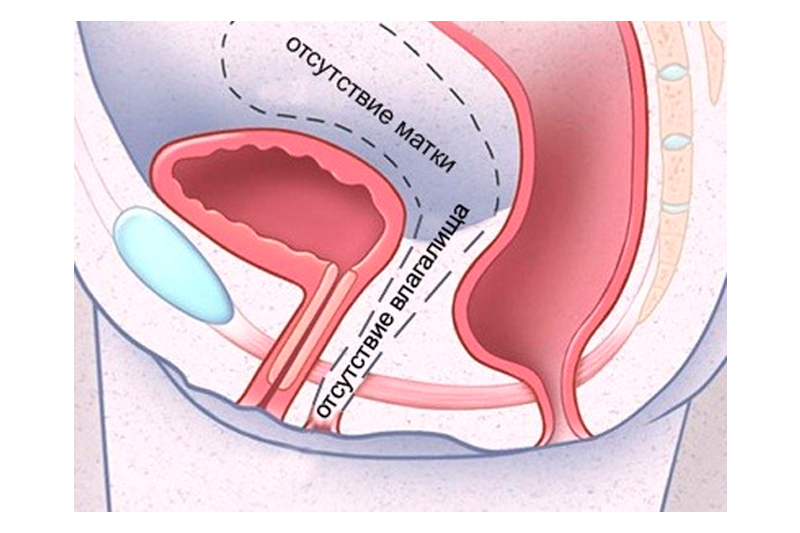
Эмбриопатическая форма развивается вследствие тератогенных факторов или развития спонтанных мутаций, что приводит к нарушению эмбриогенеза. Патогенез развития заболевания заключается в дефектах процесса формирования мюллеровых протоков, с которых развиваются женские половые пути. Органогенез женских половых путей длится с 3 по 20 неделю, и действие тератогенных факторов именно в этот период может привести к развитию синдрома Рокитанского-Кюстнера.
Симптомы синдрома Рокитанского-Кюстнера
Основным симптомом болезни Рокитанского-Кюстнера является отсутствие менструаций (аменорея). Симптом носит первичный характер, то есть у девочки не наступает менархе и в дальнейшем менструации не происходят. Первичная аменорея объясняется отсутствием матки. Чаще всего болезнь диагностируют в 14-15 лет, когда девушки обращаются к гинекологу с этой жалобой. Второй симптом — невозможность половых отношений, поскольку влагалище или отсутствует, или же определяется в виде слепо закрытого рудиментарного мешка. Известны случаи тяжелых разрывов промежности при попытке полового акта.
Частым явлением при синдроме Рокитанского-Кюстнера являются патологии развития других систем, которые в большинстве случаев диагностируются еще в детском возрасте. Очень распространены пороки мочевыводящей системы: аплазия почки и дистопия почки, подковообразная почка, раздвоения мочеводов. Симптоматика этих заболеваний разнообразна, преимущественно выражается почечной недостаточностью и дизурическими явлениями.
Диагностика заболевания
Диагностировать синдром Рокитанского-Кюстнера не составляет трудностей. Диагноз ставится уже при первом визите к гинекологу, где при осмотре на гинекологическом кресле обнаруживают слепо закрытый карман вместо влагалища, а на УЗИ малого таза визуализируют отсутствие матки (или соединительнотканный тяж) и наличие нормально функционирующих яичников. При этом телосложение пациентки сложено за женским типом с нормально развитыми молочными железами и внешними половыми органами.
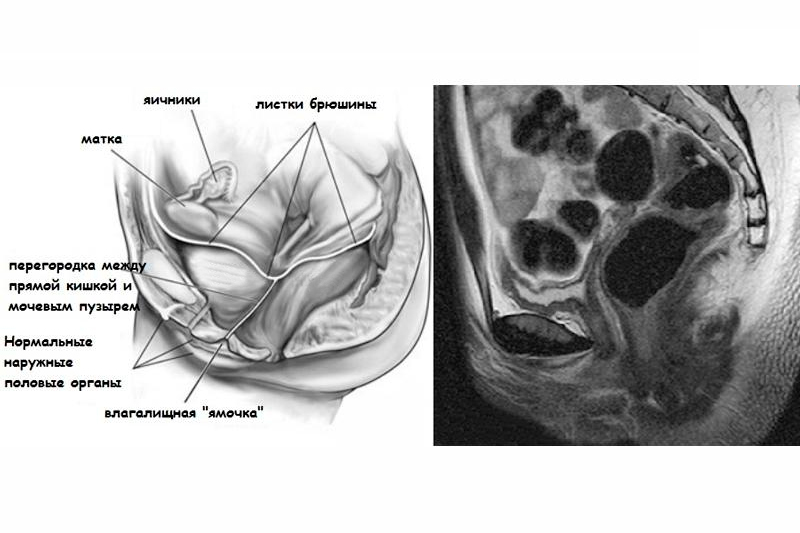
Дополнительно подтвердить синдром Рокитанского-Кюстнера позволяет определение уровня гонадотропинов и стероидов в крови, которые будут варьировать в границах нормы. Этот тест подтверждает то, что гормональная функция яичников не нарушена.
Синдром Рокитанского-Кюстнера дифференцируют с синдромом тестикулярной феминизации (синдромом Морисса), которым страдают представители мужского пола. Причиной развития этого врожденного заболевания является нарушение чувствительности андрогензависимых тканей и клеток мальчика к действию мужских половых гормонов. Дифференцировать эти две болезни помогает кариотипирование: у больных синдромом Рокитанского-Кюстнера-Майера набор хромосом 46ХХ, при синдроме тестикулярной феминизации-46ХУ.
Лечение заболевания
Поскольку синдром Рокитанского-Кюстнера-Майера-Хаузера представляет собой анатомический дефект женской половой системы, то единственным методом лечения, разработанным на сегодняшний день, является пластическая реконструктивная операция. Суть операции — в пластическом формировании неовагины, материалом для которой становятся часть сигмовидной кишки или тазовой брюшины. Выбор этих двух материалов для пластики объясняется схожестью тканей кишечника и брюшины с тканью влагалища, что отнимает потребность дополнительно увлажнять неовагину. В случае, если длина брыжейки сигмовидной кишки не позволяет провести реконструктивную операцию, материалом становятся часть тонкой или другая часть толстой кишки.
На сегодняшний день проводят два вида оперативных вмешательств: открытое и лапароскопическое. Последнее считается более рациональным, поскольку позволяет сберечь эстетичный вид брюшной стенки и снижает сроки реабилитации после вмешательства. Цель этих операций — восстановление нормальной половой жизни женщины. Неовагина в большинстве случаев функционирует нормально, однако иногда она поддается склерозированию, что приводит к стенозу просвета. Для профилактики этих явлений рекомендуют периодически проводить бужирование просвета неовагины и вести активную половую жизнь.
Операции с восстановления матки на сегодняшний день не разработаны, поэтому выносить ребенка женщина не сможет. Однако, учитывая то, что яичники у пациенток функционируют нормально, существует процедура оперативного забора яйцеклетки и последующего экстракорпорального оплодотворения с инкорпорированием в матку суррогатной матери.
Внимание! Данная статья размещена исключительно в ознакомительных целях и ни при каких обстоятельствах не является научным материалом или медицинским советом и не может служить заменой очной консультации с профессиональным врачом. За диагностикой, постановкой диагноза и назначением лечения обращайтесь к квалифицированным врачам!
Количество прочтений:
Дата публикации: 03.09.2018
Источник

Аплазия матки и влагалища (синдром Рокитанского-Кюстнера) – редкая врожденная аномалия, характеризующаяся первичным отсутствием матки и 2/3 верхних отделов влагалища. При синдроме Рокитанского-Кюстнера сохраняется нормальное развитие наружных гениталий, яичников и выраженность вторичных половых признаков, отсутствуют хромосомные аномалии (имеется женский кариотип 46ХХ). Характеризуется первичной аменореей, нередко сочетается с пороками других систем. Диагностируется в ходе гинекологического обследования (осмотра, УЗИ, МРТ, лапароскопии). Лечение синдрома Рокитанского-Кюстнера – хирургическое, сводится к созданию неовагины – искусственной влагалищной трубки.
Общие сведения
Частота встречаемости синдрома Рокитанского-Кюстнера составляет 1 на 4500-5000 случаев новорожденных девочек. Регистрируются как спорадические, так и семейные формы аномалии. Современная гинекология выделяет изолированный синдром Рокитанского-Кюстнера, характеризующийся только аплазией матки и влагалища, а также тип порока, сочетающийся с врожденными аномалиями развития позвоночника, почек, сердца и др. органов.
Первое упоминание порока развития матки и влагалища относится к началу XIX в., когда немецкий ученый Майер описал аплазию влагалища, сочетающуюся с множественными аномалиями развития. Позднее Рокитанским и Кюстером было замечено, что при данном синдроме также отсутствует матка, но сохранены и нормально функционируют яичники. В дальнейшем Хаузер обнаружил частое сочетание аплазии матки и влагалища с пороками развития почек и скелета.

Синдром Рокитанского-Кюстнера
Этиология
Обусловленность развития синдрома Рокитанского-Кюстнера во многом остается неясной. В возникновении спорадических случаев синдрома Рокитанского-Кюстнера играет роль нарушение эмбриогенеза, а именно – развития мюллеровых протоков, являющихся предшественниками женских половых органов. Эти нарушения могут быть спровоцированы недостаточной продукцией биологически активной субстанции MIS в клетках эмбриона, тератогенными факторами (гестационным диабетом, химическими агентами), дефектами формирования мезенхимы и т. д.
Эпизоды семейного синдрома Рокитанского-Кюстнера наследуются по аутосомно-доминантному типу передачи с неполной пенетрантностью и экспрессивностью гена. Синдрома Рокитанского-Кюстнера в гинекологии диагностируется у женщин с первичной аменореей в 20% случаев.
Клинические проявления
Основной жалобой, с которой девочки 15-16 лет приходят на консультацию гинеколога, служит отсутствие менструаций. Пациенток более старшего возраста обычно беспокоит невозможность ведения половой жизни. В отдельных случаях синдром Рокитанского-Кюстнера выявляется в результате экстренного обращения женщины в связи с тяжелыми разрывами промежности при попытке полового сношения.
Лица с синдромом Рокитанского-Кюстнера сложены по женскому типу, имеют нормально развитые наружные гениталии и вторичные половые признаки. При синдроме Рокитанского-Кюстнера влагалище либо полностью отсутствует, либо определяется в виде укороченного слепого мешка. Недоразвитие внутренних половых органов может проявляться наличием рудиментной матки в виде небольшого тяжа или двух рогов с тонкими неполноценными фаллопиевыми трубами или полным их отсутствием. При этом яичники имеют нормальную архитектонику и полноценно функционируют.
У 40% женщин синдром Рокитанского-Кюстнера сочетается со скелетными аномалиями и пороками мочевой системы – подковообразной почкой, дистопией почки, аплазией почки, удвоением мочеточников. Больные с синдромом Рокитанского-Кюстнера бесплодны.
Диагностика
Общий осмотр выявляет правильное телосложение, соответствие физического и полового развития возрастным нормам. При гинекологическом осмотре на кресле пациенток с синдромом Рокитанского-Кюстнера определяется нормальное развитие наружных гениталий, оволосение по женскому типу. Исследование влагалища с помощью зонда выявляет за девственной плевой укороченное и слепо заканчивающееся влагалище длиной 1-1,5 см. Ректально-абдоминальное исследование позволяет пальпировать тяж в месте типичного расположения матки и отсутствие придатков.
График измерения БТ в прямой кишке соответствует смене фаз менструального цикла и подтверждает нормальное функционирование яичников. Показатели уровня гонадотропинов и стероидов в плазме соответствуют норме. Проведение трансабдоминального УЗИ или МРТ органов малого таза удостоверяет наличие синдрома Рокитанского-Кюстнера. На УЗИ почек нередко выявляются аномалии мочевыводящих путей. Иногда для уточнения степени аплазии перед проведением кольпопоэза прибегают к диагностической лапароскопии. Синдром Рокитанского-Кюстнера дифференцируют с атрезией влагалища и матки при синдроме тестикулярной феминизации (мужской кариотип 46-XY).
Лечение
Единственным способом лечения синдрома Рокитанского-Кюстнера является пластическое формирование неовлагалища – кольпопоэз из тазовой брюшины или сигмовидной кишки. Бужирование и дилятация рудиментного влагалища (кольпоэлонгация) возможна только при его достаточной длине – 2-4 см. Целью вмешательств служит устранение препятствия для нормальной половой жизни.
В большинстве случаев в реконструктивно-восстановительной интимной пластике проводится лапароскопический кольпопоэз с ротацией фрагмента сигмовидной кишки на собственной брыжейке. Искусственное влагалище, сформированное из тканей кишки, не требует дополнительного увлажнения, что положительно сказывается на качестве сексуальной жизни женщин с синдромом Рокитанского-Кюстнера.
Ткани неовагины после операции подвергаются некоторым морфофункциональным изменениям, выражающимся в склерозе, атрофии и дисплазии слизистой кишки. Чтобы не допустить склерозирования и стеноза просвета искусственного влагалища необходимо ведение регулярной половой жизни или периодическое проведение бужирования.
Лапароскопический кольпопоэз при синдроме Рокитанского-Кюстнера имеет преимущества перед открытым вмешательством, позволяя достичь лучших эстетических результатов. При невозможности выполнения кольпопоэза из сигмовидной кишки (недостаточной длине брыжейки) для пластики неовлагалища используются тазовая брюшина, поперечно-ободочная или тонкая кишка.
Прогноз
При синдроме Рокитанского-Кюстнера возможен только паллиативный вариант лечения, которое направлено на адаптацию пациентки к нормальной сексуальной жизни при сохранении аменореи. После пластики влагалища у пациенток с синдромом Рокитанского-Кюстнера нормализуется половая функция. Рождение генетических детей от женщин с синдромом Рокитанского-Кюстнера может быть достигнуто только с использованием экстракорпорального оплодотворения и суррогатного материнства.
Синдром Рокитанского-Кюстнера – лечение в Москве
Источник