
Синдром шерешевского тернера и клайнфельтера примеры

Синдром Клайнфельтера и Шерешевского — Тернера. Гермафродитизм
Синдром Клайнфельтера наблюдается у лиц с мужским фенотипом. Характерной особенностью, объединяющей индивидуумов с различным набором половых хромосом в одну группу, является недоразвитие яичек и отсутствие сперматогенеза. Больные с синдромом Клайнфельтера обычно имеют высокий рост, евнухоидные пропорции тела (узкие плечи, широкий таз), склонность к ожирению, иногда гинекомастию (одно- или двустороннюю), оволосение по женскому типу. Основная масса таких больных имеет кариотип XXY.
Описаны также больные с набором половых хромосом XXXY, XXXXY, XXYY и др. При исследовании интерфазных ядер у таких больных обнаруживают как Х-, так и Y-хроматин, причем число телец Х-хроматина равно числу Х-хромосом минус единица. Так, например, у индивидуума с кариотипом XXXXY в ядрах некоторых клеток может быть обнаружено до 3 телец Х-хроматина и одно Y-хроматина.
Синдром Шерешевского — Тернера наблюдается у лиц фенотнпически женского пола с задержкой роста и полового развития, нормальными наружными и недоразвитыми внутренними половыми органами. Наиболее характерными чертами являются отсутствие гонад, первичная аменорея, бесплодие, короткая шея и крыловидные складки кожи на шее.
Женщины с синдромом Тернера имеют кариотип ХО, Х-хроматин в клетках их тканей отсутствует.
Ложный мужской гермафродитизм. У больных имеются тестикулы, а дифференциация других внутренних и всех или некоторых наружных половых органов отклоняется в женскую сторону. Разновидностью этого состояния является аномалия полового развития, называемая тестикулярной феминизацией, проявляющаяся присутствием тестикул у фенотипических женщин с первичной аменореей. Отмечается нормальное, часто чрезмерное развитие грудн. Наружные половые органы женские, внутренние отсутствуют. Кариотип этих больных содержит XY-хромосомы.
В ядрах клеток Х-хроматин отсутствует, а Y-хроматин обнаруживается.
Ложный женский гермафродитизм — аномалия полового развития, несвязанная с нарушением в системе половых хромосом. Заболевание обусловлено врожденной гиперплазией коры надпочечников. Основные клинические проявления синдрома вызываются избытком мужских гормонов — андрогенов, обусловливающих вирилизирующий эффект. Все больные имеют Х-хроматин и нормальный женский кариотип.
Истинный гермафродитизм. Под этим названием понимают такое интерсексуальное состояние, когда у индивидуума одновременно имеются две половые железы — мужского и женского пола (яичко и яичник) или железы, состоящие из тканей мужской и женской половых желез (овотестис). Строение наружных половых органов характеризуется различной степенью переходов от одного пола к другому. Около 60% описанных случаев имели ХХ-хромосомную конституцию. В 40% случаев в кариотипе имелась и Y-хромосома.
Полисомия по Х-хромосоме у женщин (XXX, ХХХХ, ХХХХХ) характеризуется наличием в некоторых интерфазных ядрах двух — трех телец Х-хроматнна. Это пограничное между патологией и нормой состояние сопровождается эндокринным дисбалансом и в первую очередь нарушением функции яичников. У части обследованных женщин выявлены бесплодие, нерегулярность менструального цикла, преждевременный климакс.
Аномальные наборы половых хромосом могут возникать не только в процессе гамстогенеза (при нарушении распределения их в митозе), но и в процессе эмбрионального развития (при нарушении распределения их в анафазе митотического деления). Последние нарушения приводят к появлению линий (клонов) клеток, отличающихся от исходных; формируется мозаичный (по половым хромосомам) организм.
Например, 46, XY/47, XXY — наиболее частый тип мозаичности по половым хромосомам у мужчин, и 45, Х/46, XX — у женщин. Процентное содержание клеток разных клонов, распределение их в тканях организма зависит от стадии эмбрионального развития, на которой появилась аномалия, и от выживаемости аномального клона. В зависимости от преобладания клеток того или иного клона варьируют и клинические проявления.
Истинную природу интерсексуальных заболеваний можно выяснить только при сопоставлении результатов клинического осмотра и лабораторных исследований. Большую ценность для диагностики представляет установление кариотипа больного и комплекса половых хромосом. Число и состав половых хромосом можно определить путем исследования Х- и Y-хроматина, так как между числом половых хромосом и числом телец полового хроматина в покоящемся ядре существует вполне определенное соотношение.
– Также рекомендуем “Определение половой принадлежности материалов. Половая принадлежность сред организма”
Оглавление темы “Определение половой принадлежности клеток”:
1. Содержание Y-хроматина в клетках. Аномалии содержания полового хроматина
2. Синдром Клайнфельтера и Шерешевского — Тернера. Гермафродитизм
3. Определение половой принадлежности материалов. Половая принадлежность сред организма
4. Половая принадлежность по Х-хроматину. Определение пола по Х-хроматину
5. Приготовление препаратов крови для выявления Х-хроматина. Выявление пола по крови и слюны
6. Микроскопия для определения пола. Исследование препаратов для определения пола клеток
7. Микроскопия крови, волос для определения пола. Оценка результатов выявления пола
8. Статистический метод определения пола. Последовательный анализ Вальда
9. Половая принадлежность Y-хроматину. Приготовление препаратов для определения пола по Y-хроматину
10. Микроскопия для выявления Y-хроматина. Исследование препаратов на Y-хроматин
Источник
Оглавление
- Введение
- Краткое содержание
- Аномалии числа хромосом (анеуплоидии)
- Моносомия по X-хромосоме (45,X, или Синдром Шерешевского-Тёрнера)
- 47,XXY Синдром Клайнфельтера
- 47,XYY
- 47,XXX
- Другие заболевания
- Мозаицизм 45,X/46,XX
- Мозаицизм 45,X/46,XY
- Структурные аномалии хромосом
- Изохромосома Xq
- Делеция Xp22.11
- Делеция Xp22.3
- Делеции Xp22 SHOX
- Делеции Xp11.22
- Дупликации Xp.22.31
- Синдром дупликации ME2CP
Введение
Патологии половых хромосом связаны с нарушением их количества (т. е. анеуплоидии, например, моносомия X-хромосомы) или со структурными дефектами (например, такие геномные перестройки, как синдром дупликации гена MECP2). Частота врожденных хромосомных мутаций составляет как минимум 1:400.
Краткое содержание
Патологии половых хромосом могут быть обусловлены нарушением их количества (анеуплоидиями) или же структурными дефектами.
Наиболее распространенные анеуплоидии половых хромосом: 45,X (Синдром Тёрнера); 47,XXY (Синдром Клайнфельтера); 47,XYY; и 47,XXX. Мозаицизм по половым хромосомам с присутствием в организме клеток с нормальным генотипом нередок. Два наиболее распространенных вида мозаицизма половых хромосом — 45,X/46,XX и 45,X/46,XY. Тяжесть фенотипических проявлений у пациентов с мозаицизмом соответствует доле аномальных клеток.
Структурные патологии X- и Y-хромосом прежде всего включают изохромосомы, делеции, дупликации, кольцевые хромосомы и транслокации.
Одним из примеров геномного расстройства является дупликация гена MECP2 у мужчин, выражающаяся в наличии мышечной гипотонии, тяжелой умственной отсталости, задержки речевого развития, нарушения глотания, частых респираторных инфекций, а также судорожных приступов (тонико-клонические судороги, не поддающихся лечению).
Аномалии числа хромосом (анеуплоидии)
Наиболее частыми анеуплоидиями половых хромосом являются 45,X (Синдром Шерешевского-Тернера); 47,XXY (Синдром Клайнфельтера); 47,XYY и 47,XXX с частотой возникновения приблизительно 1/2500, от 1/500 до 1/1000, от 1/900 до 1500 и 1/1000 соответственно. Мозаицизм по половым хромосомам с присутствием в организме клеток с нормальным генотипом нередок. Два наиболее распространенных вида мозаицизма половых хромосом — 45,X/46,XX и 45,X/46,XY. Тяжесть фенотипических проявлений у пациентов с мозаицизмом соответствует проценту аномальных клеток.
Моносомия по X-хромосоме (45,X, или Синдром Шерешевского-Тёрнера)
Большинство пациентов с синдромом Шерешевского-Тёрнера имеют моносомию по Х-хромосоме, кариотип 45,X. Другие формы синдрома включают мозаицизм по хромосоме Х, например, 45,X/46,XX или 45,X/46,XY с частичной делецией Y-хромосомы. У некоторых пациентов имеется структурная аномалия второй X-хромосомы (например, изохромосомия длинного плеча X-хромосомы или делеция короткого плеча). Делеции, включающие в себя дистальную часть короткого плеча Y-хромосомы, также ассоциированы с фенотипом синдрома Тёрнера, поскольку в данном случае у пациентов отсутствуют так называемые анти-тёрнеровские гены (SHOX, RPSY4 и ZFY). Делеции короткого плеча X-хромосомы также связывают с фенотипом синдрома Тёрнера. В большинстве представляют собой единичные случаи.
Синдром Шерешевского-Тёрнера характеризуется низкорослостью и некоторыми из следующих проявлений: дисморфия лица, включающая низко посаженные уши, кожные складки на шее, щитообразная грудная клетка (широкая, с большим расстоянием между сосками), лимфедема, вальгусная деформация локтевого сустава, короткая четвертая пястная кость, гипоплазия ногтевых пластин, пигментные пятна и врожденные пороки сердца. Среди пороков сердца типичными и наиболее часто встречающимся являются дефекты сосудов и коарктация аорты. Вдобавок у пациентов, страдающих синдромом Тёрнера, развиваются полосковидные гонады, наблюдается нарушение овуляции и задержка полового развития. Также встречаются дефекты развития почек (подковообразная почка). Лимфедема нижних отделов конечностей может быть единственным клиническим признаком, наблюдаемым у новорожденных. Лица с синдромом Тёрнера, несущие генетический материал Y-хромосомы, имеют повышенный риск развития гонадобластомы.
47,XXY Синдром Клайнфельтера
Синдром Клайнфельтера является самой распространенной патологией числа половых хромосом, вызывающей первичный гипогонадизм. Кариотип 47,XXY является результатом нерасхождения половых хромосом и может быть как материнским, так и отцовским по происхождению. Большинство случаев болезни обнаруживается постнатально и диагностируется при определении причин бесплодия, выявлении гинекомастии, крипторхизма или же неврологических нарушений.
Рис. Нерасхождение половых хромосом
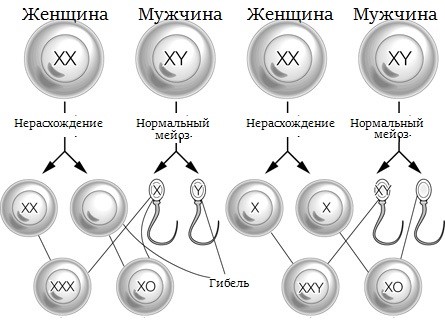
Новорожденные мальчики с кариотипом 47,XXY фенотипически нормальны, с физиологически нормальными мужскими наружными половыми органами и без какой-либо видимой дисморфии. Основные клинические проявления синдрома Клайнфельтера, включающие высокий рост, маленькие яички и бесплодие (азооспермия), становятся выраженными в постпубертатном периоде. У пациентов с синдромом Клайнфельтера повышен риск психических расстройств, расстройств аутистического характера и социальных проблем. У пациентов с диагностированным синдромом Клайнфельтера следует оценивать неврологический статус и направлять к эндокринологу.
47,XYY
Лица с кариотипом 47,XYY имеют высокий рост, у них может отмечаться умеренная задержка в двигательном и речевом развитии. Для многих из них требуется повышенное внимание к обучению, но, как правило, все они учатся в основных общеобразовательных школах. Половое развитие проходит нормально и большинство мальчиков фертильны. Из-за невыраженности фенотипа и отсутствия связанных с этим проблем со здоровьем, многие лица с кариотипом 47,XYY на протяжении всей их жизни остаются недиагностированными.
Ранее сообщалось, что у мужчин с 47,XYY повышена агрессия, что выражается в их агрессивном поведении. Однако последующие крупномасштабные совместные исследования европейских и американских генетиков показали, что статистика повышенной криминальной деятельности мужчин с XYY коррелировала с их низким социально-экономическим статусом по причине низкого значения IQ (около 10 баллов), что приводило к определенным трудностям с законом и, чаще, незначительным правонарушениям. У лиц с 47,XYY отмечаются более высокие показатели синдрома дефицита внимания и гиперактивности, а также расстройств аутистического характера. Таким пациентам рекомендуется оценка их нервно-психического развития, учитывая широкую распространённость трудностей в обучении и поведенческих проблем.
47,XXX
47,XXX (она же трисомия по X-хромосоме) является самой распространенной патологией половых хромосом у женщин. Трисомия по Х-хромосоме диагностируется внутриутробно в ходе генетического скрининга. У женщин с кариотипом 47,XXX нет повышенного риска развития плода с хромосомными аномалиями.
Обследование 155 женщин с кариотипом 47,XXX показало, что 62 процента из них были физически нормальными. Таким образом, для большинства лиц с кариотипом 47,XXX диагноз никогда не устанавливается. У женщин с 47,XXX отмечается высокий рост; (средняя длина окружности головы варьирует в пределах 25 – 35 процентиль, однако к подростковому возрасту для многих может достигать 80 процентиль). Половозрелость и фертильность чаще всего в норме, но может отмечаться преждевременное угасание функции яичников.
В следующем обследовании одиннадцати младенцев с кариотипом 47,XXX было показано, что коэффициент интеллекта девочек с рождения был на 15-20 баллов ниже, чем у их братьев. Поэтому рекомендуется отслеживать задержки в развитии и выявлять наличие психологических проблем в дальнейшем.
Другие заболевания
Сообщалось о более чем ста случаях кариотипа 49,XXXXY, по меньшей мере двадцати случаях 49,XXXXX и нескольких – 49,XYYYY. Прослеживается прямая зависимость между числом дополнительных половых хромосом и тяжестью фенотипических проявлений у пациентов. В исследовании тетра- и пентасомии половых хромосом сделан вывод о том, что полисомия по X-хромосоме связана с более тяжкими последствиями, чем полисомия по Y-хромосоме. Было показано, что уровень интеллекта IQ снижается на 10 пунктов с каждой лишней X-хромосомой от их нормального числа.
49,XXXXY Характерными клиническими чертами кариотипа XXXXY являются запавшая переносица с широким или приподнятым кончиком носа, широко расположенные глаза, веко-носовые складки, скелетные патологии (особенно лучелоктевой синостоз), врожденные сердечные заболевания, эндокринные расстройства и высокая степень гипогонадизма и гипогенитализма. Также обычным являются выраженная умственная отсталость и умеренная низкорослость. Хотя лиц с таким кариотипом часто относят к случаям синдрома Клайнфельтера, все характерные черты XXXXY довольно отчетливо указывают именно на данный фенотип.
49,XXXXX У женщин с кариотипом 49,XXXXX (пентасомия по X-хромосоме) всегда присутствует умственная отсталость. Другие проявления, такие как черпено-лицевые, сердечно-сосудистые и скелетные патологии довольно непостоянны. У пациентов, страдающих пентасомией по X-хромосоме, могут проявляться схожие черты с теми, что наблюдаются при синдроме Дауна. Лучелоктевой синостоз также часто выражен у пациентов с большим числом X-хромосом. Некоторые пациенты имеют мозаицизм 48,XXXX и 49,XXXXX.
Мозаицизм 45,X/46,XX
Это наиболее распространенный мозаицизм половых хромосом, который диагностируется при амниоцентезе и пренатальном кариотипировании. У лиц с данным типом мозаицизма имеются более легкие клинические черты синдрома Тёрнера. Многие женщины прошли половое созревание и смогли воспроизвести потомство.
Из 156 пренатально диагностированных случаев мозаицизма 45,X/46,XX 14% случаев имели ненормальный исход. Было зарегистрировано два мертворождения и 20 случаев ненормального фенотипа (у 12 имелись некоторые черты синдрома Тёрнера, а остальные 8 носили характер аномалий, возможно, не связанных с ним). Более 85 % девочек имели нормальный фенотип при рождении, либо он был установлен по результатам медицинского прерывания беременности. Однако, главные черты синдрома Тёрнера (такие как низкий рост и отсутствие вторичных половых признаков) проявились только в детстве или юности, и не были замечены в младенчестве. У некоторых женщин с нормальным фенотипом, при нарушении функции яичников, выявляется мозаицизм 45,X/46,XX.
Мозаицизм 45,X/46,XY
Мозаицизм с наличием 45,X/46,XY имеет широкий фенотипический спектр. Например, в ретроспективной серии 151 постнатально диагностированных случаев мозаицизма 45,X/46,XY, 42 % пациентов — девочки по фенотипу, с наличием типичного или нетипичного синдрома Тёрнера. Еще у 42 % наблюдались неопределённые наружные половые органы и асимметричные гонады (смешанный гонадный дисгенез), наконец, у 15% был мужской фенотип с неполной маскулинизацией. Таким образом, все случаи, диагностированные постнатально, были фенотипически патологичными. Напротив, среди 80 пренатально диагностированных случаев мозаицизма 45,X/46,XY 74 92,6% были нормальными по фенотипу мальчиками. Это может объяснить тот факт, что дети или взрослые с наличием мозаицизма, но нормальным фенотипом вряд ли стали бы обращаться за медицинской помощью (ошибка обращаемости).
Структурные аномалии хромосом
Структурные патологии включают, прежде всего, изохромосомы, делеции, дупликации, кольцевые хромосомы и транслокации.
Изохромосома Xq
Изохромосома длинного плеча X-хромосомы, isoXq или i(Xq), при наличии которой короткое плечо (p) исключено (отсутствует/редуцировано) и заменено точной копией длинного плеча (q), — является наиболее распространенной аномалией половых хромосом.
Наличие структурной патологии не связывают с повышенным возрастным риском родителей. Изохромосомия 46,X,i(Xq) может быть выражением мозаицизма, когда в организме присутствуют две генетически разные клеточные популяции: нормальная – 46,XX и 45,X.
Изохромосомы Xq и Xy ассоциируют с синдромом Тёрнера, возможно, потому, что главный анти-тёрнеровский ген SHOX располагается на дистальной части коротких плеч X-и Y-хромосом (на псевдоаутосомных областях). Изохромосома Xq также выявляется у пациентов в одной из вариаций синдрома Клайнфельтера, 47,X,i(Xq),Y.
Делеция Xp22.11
Делеция Xp22.11 включает в себя ген PTCHD1. Сообщалось о выявлении в нескольких семьях с расстройствами аутистического характера, а также в трёх семьях с умственной отсталостью. Ген PTCHD1 является геном-кандидатом в отношении Х-сцепленной умственной отсталости, проявляющейся с аутизмом или без аутизма. Функция и роль данного гена неизвестны.
Делеция Xp22.3
Делеция данной области часто ассоциируется с синдромом микрофтальмии и линейных дефектов кожи (MLS) и является Х-сцепленным доминантным нарушением, то есть, летальным для мужчин и поэтому прослеживающимся только у женщин. Ген в данной области кодирует митохондриальную цитохром-c-синтазу (HCCS). Клиническое проявление MLS выражается наличием микрофтальмии и анофтальмии (одно- или двусторонней) и линейными дефектами кожи, в основном лица и шеи, которые со временем проходят. Структурные патологии головного мозга, задержка в развитии и приступы (припадки) тоже входят в состав клинической картины. Нарушения сердечной деятельности (как гипертоническая кардиомиопатия и аритмия), низкий рост, грыжа диафрагмы, ногтевая дистрофия, преаурикулярный свищ, потеря слуха, мочеполовые мальформации (пороки развития, неправильное формирование) также являются частыми клиническими явлениями.
Скрининговая оценка предусматривает офтальмологический и дерматологический осмотр, оценку общего развития, выполнение эхокардиограммы, магнитно-резонансной томографии мозга (МРТ) и электроэнцефалограммы (ЭЭГ).
Делеции Xp22 SHOX
Делеция Xp22 включает в себя ген SHOX, мутация которого является причиной идиопатического низкого роста. Ген SHOX находится в псевдоаутосомном регионе 1 X- и Y-хромосом. Этот ген считается ответственным за низкорослость при синдроме Тёрнера, а гаплонедостаточность данного гена вызывает дисхондростеоз Лери-Вейлля. Дисхондростеоз Лери-Вейлля характеризуется низким ростом, наиболее выражено проявляющимся у женщин, а также хроническим подвывихом кисти (деформацией костей запястья, деформация Маделунга). Гомозиготные делеции гена SHOX вызывают дисплазию Лангера, более тяжелую форму метафизарной дисплазии. Делеции гена SHOX легко обнаруживаются у пациентов с низким ростом, без каких-либо других специфических особенностей в строении их скелета. Более чем 60% перестроек SHOX – это делеции гена; при отсутствии делеций сравнительная геномная гибридизация с последующим секвенированием для выявления и установления точечных мутаций, является клиническим обследованием идиопатического низкого роста.
Делеции Xp11.22
Делеции региона Xp11.22 включают ген PHF8 (кодирует пальцевидный белок PHD8), мутации которого связывают с умственной отсталостью, наличием расщелины губы/неба, а также с расстройствами аутистического характера.
Мутации с делецией гена PHF8 ассоциированы с синдромом Х-сцепленной умственной отсталости, синдром Сидериус-Хамель (синдром Siderius-Hamel).
Дупликации Xp.22.31
Дупликации в локусе Xp.22.31 часто описываются в литературе. Было много дискуссий на тему того, является ли данная дупликация патогенетической или же доброкачественным явлением, учитывая трудности определения последствий вариации числа копий генов. Данная дупликация затрагивает ген стероидной сульфатазы. Как результат – генетический дефект, мутация в гене стероидной сульфатазы, что выражается в снижении её активности или отсутствие её синтеза. Делеция данного гена связана с Х-сцепленным ихтиозом у мужчин. Данная дупликация отмечается у пациентов с умственной отсталостью. Однако, она выявляется как у здоровых родственников этих пациентов, так и в основной популяции. Хотя дупликации данного гена могут и не иметь фенотипических проявлений, трипликации последовательно связывают с умственными расстройствами. FISH-диагностика позволяет в конечном счете дифференцировать дупликации от трипликации (распознать увеличение копийности гена).
Синдром дупликации ME2CP
Мутации в гене, кодирующем метил-связывающий-CpG терминальный белок 2 (ME2CP), расположенный в Xq28, ответственный за синдром Ретта. Дупликации данного региона имеет небольшое или вовсе не имеет фенотипического значения для женщин, вероятно, из-за инактивации патологической X-хромосомы. Мужчины с данной мутацией сильно ослаблены. Наличие дупликации клинически выражается в наличии выраженной мышечной гипотонии, тяжелой умственной отсталости, задержке речевого развития, нарушения глотания (трудностей приема пищи), частых респираторных инфекций и судорожных приступов вплоть до тонико-клонических, иногда не поддающихся лечению. Многие пациенты с наличием данной дупликации были с диагностированным аутизмом либо расстройством подобного типа. По аналогии с тем, что наблюдается в синдроме Ретта, пациенты с дупликацией ME2CP испытывают регресс развития. Вдобавок у них развивается атаксия, прогрессирующие мышечные спастичности нижней части тела часто приводят к потере способности передвигаться. Отмечались проблемы желудочно-кишечного тракта и сильные запоры. Дупликация часто затрагивает ген антагонист рецептора интерлейкина 1 (IRAK1), что может играть роль в появлении иммунных патологий, отмечаемой у данной группы пациентов. Прогноз неблагоприятен, и большинство мужчин с данной дупликацией умирают до 30 лет по причине вторичных респираторных инфекций. Трипликация данного региона проявляется еще более тяжелым фенотипом у мужчин.
Скрининговые обследования этих пациентов предполагают проведение ЭЭГ, оценку функции глотания, оценку гуморального и клеточного иммунитета. Лечение может включать лечение мышечной гипотонии и спастичности, речевую терапию (логопедию), использование гастрономической трубки (гастростома) в случае проблем с питанием, а также лечение респираторных инфекций.
Перевод материалов сайта UpTodate подготовлен специалистами Центра иммунологии и репродукции.
Источник